مقدمه
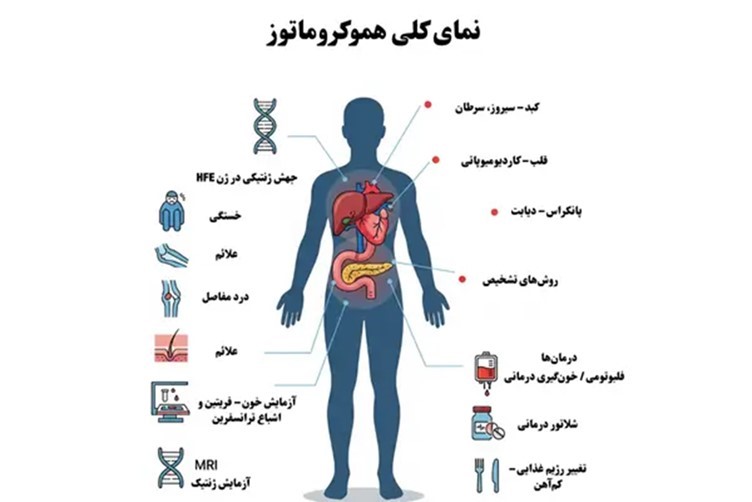
هموکروماتوز ارثی (Hereditary Hemochromatosis یا HH) یکی از شایعترین اختلالات ژنتیکی متابولیسم آهن است که با جذب بیش از حد آهن از دستگاه گوارش و تجمع آن در بافتهای بدن همراه است. این بیماری که عمدتاً بر اندامهایی مانند کبد، قلب، پانکراس و غدد درونریز تأثیر میگذارد، میتواند منجر به عوارض جدی مانند سیروز کبدی، نارسایی قلبی، دیابت و حتی سرطان کبد شود. بر اساس بررسیهای اخیر، شیوع HH در جمعیتهای اروپایی شمالی تا ۱ در ۲۰۰-۳۰۰ نفر میرسد، در حالی که این بیماری در ایران و خاورمیانه کمتر شایع است اما گمان می کنیم که با مهاجرتها و غربالگری ژنتیکی، موارد بیشتری شناسایی شود. تشخیص زودهنگام و درمان مناسب میتواند از پیشرفت بیماری جلوگیری کند و کیفیت زندگی را حفظ نماید.
پاتوفیزیولوژی
پاتوفیزیولوژی HH بر پایه اختلال در هموستازی آهن استوار است. هورمون هپسیدین که توسط کبد ترشح میشود، نقش اصلی در تنظیم جذب آهن از دوازدهه روده ایفا میکند. در حالت طبیعی، هپسیدین با اتصال به فروپورتین (کانال خروجی آهن از سلولهای رودهای)، جذب آهن را محدود میسازد. در HH، جهشهای ژنتیکی منجر به کاهش تولید یا عملکرد هپسیدین میشود که نتیجه آن جذب مداوم ۲-۳ برابر آهن طبیعی (تا ۴-۵ میلیگرم در روز به جای ۱-۲ میلیگرم) است. آهن اضافی به صورت فروتین در ماکروفاژها و سپس به صورت هموسیدرین در سلولهای پارانشیمی (مانند هپاتوسیتها) ذخیره میشود. این تجمع، استرس اکسیداتیو ایجاد کرده و از طریق تولید رادیکالهای آزاد، آسیب سلولی، التهاب و فیبروز را به دنبال دارد. در کبد، این فرآیند به فیبروز و سیروز منجر میشود؛ در قلب، به کاردیومیوپاتی و آریتمی؛ و در پانکراس، به دیابت برنز (bronze diabetes) به دلیل آسیب به سلولهای بتا.
بر اساس مطالعات اخیر (1403)، حتی در افراد با سطوح آهن طبیعی اما ژنوتیپ پرخطر، ریسک افزایشی مرگومیر مشاهده شده که نشاندهنده نقش عوامل محیطی مانند الکل و رژیم غذایی پرآهن است.
اساس ژنتیکی و نحوه توارث
HH عمدتاً به دلیل جهش در ژن HFE (واقع بر کروموزوم ۶) ایجاد میشود که پروتئینی را کد میکند که با گیرنده ترانسفرین تعامل دارد و سیگنالینگ هپسیدین را تنظیم مینماید. جهش شایع C282Y (جایگزینی سیستئین با تیروزین در موقعیت ۲۸۲) در هموزیگوتها نفوذ بالاتری (تا ۸۰-۹۰ درصد) دارد، در حالی که H63D و S65C نفوذ کمتری نشان میدهند. انواع نادرتر شامل نوع ۲ (جهش در HJV یا HAMP، juvenile)، نوع ۳ (TFR2) و نوع ۴ (SLC40A1، ferroportinopathy) هستند.
| نوع HH | الگوی توارث | ژن درگیر | ویژگیهای بالینی |
| نوع1 (کلاسیک) | اتوزومال مغلوب | HFE (C282Y) | تجمع آهسته، علائم پس از ۴۰ سالگی |
| نوع A2 | اتوزومال مغلوب | HJV | juvenile، شدید، قلبی-غددی |
| نوع B2 | اتوزومال مغلوب | HAMP | مشابه نوع ۲A |
| نوع 3 | اتوزومال مغلوب | TFR2 | شبیه نوع ۱ اما شدیدتر |
| نوع 4 | اتوزومال غالب | SLC40A1 | تجمع در ماکروفاژها، کمخونی |
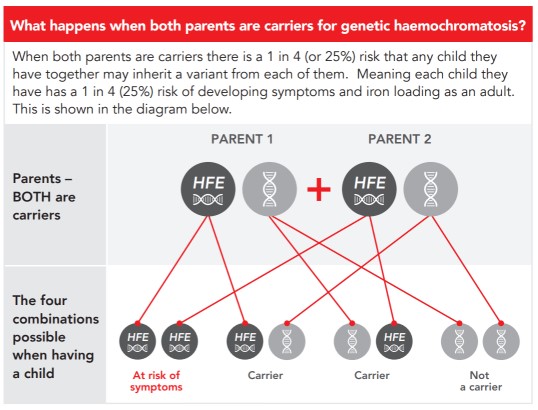
توارث HH اتوزومال مغلوب است؛ یعنی فرد باید دو آلل جهشیافته (یکی از هر والد) دریافت کند تا بیماری ظاهر شود. والدین حامل (هتروزیگوت) بدون علامت هستند، اما ریسک ۲۵ درصد برای فرزندان در انتقال دو آلل وجود دارد. نفوذ بالینی ناقص است (تنها ۲۰-۵۰ درصد هموزیگوتها علائم نشان میدهند) که تحت تأثیر جنسیت (مردان بیشتر)، سن و عوامل محیطی است.
علایم بالینی
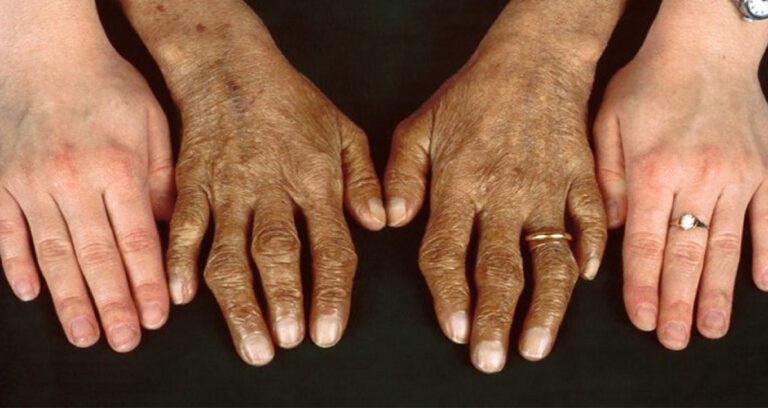
علایم HH تدریجی ظاهر میشوند و اغلب با بیماریهای دیگر همپوشانی دارند. در مراحل اولیه، خستگی مزمن و ضعف شایع است. با پیشرفت، آسیب اندامی رخ میدهد: درد مفاصل (آرتریت، به ویژه در انگشتان و زانوها)، هیپرپیگمانتاسیون پوست (برنزه شدن)، اختلالات قلبی (نارسایی احتقانی)، دیابت، هیپوگنادیسم (کاهش میل جنسی، آمنوره در زنان) و اختلالات تیروئیدی. در مردان علایم زودتر (پس از ۴۰ سال) و شدیدتر است، زیرا زنان از طریق قاعدگی آهن دفع میکنند.
| دسته علایم | مثالها |
| عمومی | خستگی، ضعف، کاهش وزن |
| کبدی | هپاتومگالی، سیروز، HCC |
| قلبی | آریتمی، کاردیومیوپاتی |
| غددی | دیابت، هیپوگنادیسم |
| اسکلتی | آرتریت، درد مفاصل |
| پوستی | هیپرپیگمانتاسیون |
تشخیص آزمایشگاهی و ژنتیکی
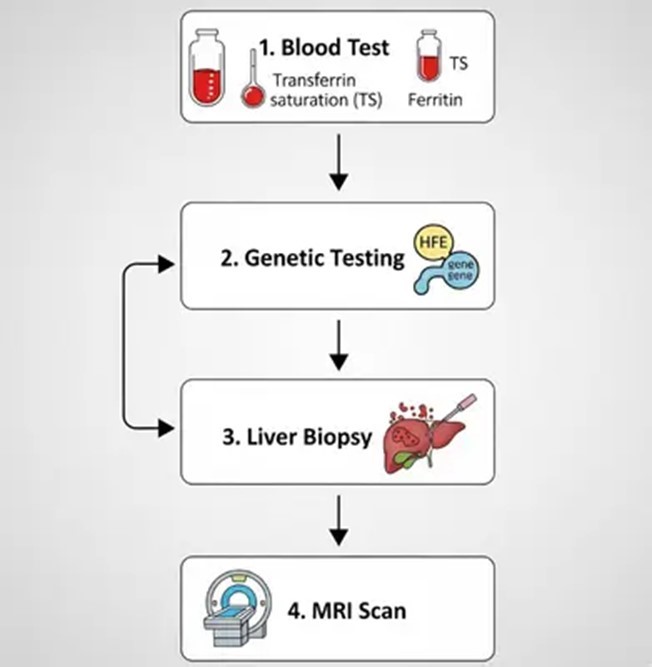
تشخیص با استفاده از ترکیبی از آزمایشهای بیوشیمیایی و ژنتیکی انجام میشود. در ابتدا، اشباع ترانسفرین (TSAT بیش از ۴۵ درصد در مردان و بیش از ۳۸ درصد در زنان) و سطح فریتین سرم (بیش از ۳۰۰ میکروگرم در لیتر در مردان و بیش از ۲۰۰ میکروگرم در لیتر در زنان) مورد بررسی قرار میگیرد. TSAT بهعنوان حساسترین نشانگر اولیه شناخته میشود. در صورت وجود شک، آزمایش ژنتیکی HFE (با استفاده از PCR برای جهشهای C282Y و H63D) توصیه میشود و وجود هموزیگوت C282Y تشخیص را تأیید میکند. در موارد نادر، انجام پنل ژنتیکی گسترده مانند پنل Invitae برای شناسایی مفید خواهد بود. همچنین، بیوپسی کبد برای ارزیابی فیبروز و MRI کبد برای کمیسازی دقیقتر استفاده میشوند. در شهر قم، آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی بوعلی خدمات مرتبط با آزمایشهای ژنتیکی را ارائه میدهد.
مشاوره ژنتیک
مشاوره ژنتیک برای افراد مبتلا، خانوادههایشان و حاملان ضروری است. شامل ارزیابی ریسک خانوادگی، توضیح نفوذ ناقص و توصیه غربالگری (آزمایش ژنتیکی برای بستگان درجه اول) میشود.
در خانوادهها، فرزندان و خواهر/برادران باید از ۱۸ سالگی غربال شوند. مشاوره پیش از بارداری برای زوجهای حامل، گزینههای غربالگری جنینی را بررسی میکند در قم، آزمایشگاهی پاتوبیولوژی و ژنتیک پزشکی بوعلی قم، این خدمات را ارائه میدهد.
راههای درمان
درمان اصلی بیماری هموکروماتوز، کاهش بار آهن از طریق فلبوتومی است که شامل دو مرحله میشود: مرحله القایی، که در آن هفتهای 450-500 میلیلیتر خون گرفته میشود تا سطح فریتین به کمتر از 50 میکروگرم بر لیتر برسد، و مرحله نگهداری، که هر 2 تا 3 ماه یک بار انجام میگیرد. بر اساس دستورالعملهای جدید ASH، فلبوتومی روشی ایمن و مؤثر بوده و احتمال عوارض را تا 90 درصد کاهش میدهد. در مواردی که بیمار قادر به تحمل فلبوتومی نیست، مانند کمخونی، از داروهای شلاتور آهن نظیر دفریپرون یا دفروکسامین استفاده میشود. رژیم غذایی کمآهن شامل اجتناب از مصرف گوشت قرمز، ویتامین C اضافی و الکل همراه با رژیم مدیترانهای نیز توصیه شده است. در ایران، فلبوتومی در مراکز نگهداری خون و بیمارستانها بهطور گسترده انجام میشود. مطالعات محلی مانند تحقیقاتی انجامشده در صدر تبریز بر خونگیری منظم و پایش سطح فریتین تأکید دارند؛ همچنین دسترسی به داروهای شلاتور آهن برای بیماران مبتلا به تالاسمی ثانویه فراهم است. درمانهای نوین مانند مینیمایزینگ هپسیدین در آزمایشهای بالینی نتایج امیدوارکنندهای نشان دادهاند، اما هنوز به عنوان استاندارد درمان شناخته نشدهاند.
پایش مادامالعمر برای جلوگیری از کارسینومای سلولهای کبدی (HCC) از طریق انجام سونوگرافی سالانه ضروری است. با تشخیص زودهنگام و مدیریت مناسب، هموکروماتوز قابل کنترل خواهد بود. غربالگری خانوادگی و مشاوره ژنتیک نیز نقش کلیدی در پیشگیری از این بیماری دارند.
Reference:
https://pubmed.ncbi.nlm.nih.gov/38980801
https://liebertpub.com/doi/full/10.1089/gtmb.2023.0764?doi=10.1089/gtmb.2023.0764
https://www.ncbi.nlm.nih.gov/books/NBK1440/
https://www.researchgate.net/publication/7132707_Hereditary_hemochromatosis_A_rare_disease_in_Iran
https://www.sciencedirect.com/science/article/abs/pii/S0025775324003919
https://pmc.ncbi.nlm.nih.gov/articles/PMC3560564/