مقدمه
سندرم زلوگر یکی از جدیترین اختلالات مرتبط با بیوژنز پراکسیزومها محسوب میشود و بهعنوان یک بیماری ژنتیکی نادر و ارثی، چندین سیستم بدن را تحت تأثیر قرار میدهد.
این عارضه که بخشی از مجموعه اختلالات طیف زلوگر (Spectrum Disorders Zellweger یا ZSD) بوده و ناشی از جهشهای اتوزومال مغلوب در ژنهای PEX است.
پراکسیزومها اندامکهای حیاتی سلولی هستند که در فرآیندهایی نظیر متابولیسم اسیدهای چرب زنجیره بلند (VLCFAs)، سنتز پلاسمالوژنها (از ترکیبات مهم غشاهای سلولی و میلین)، کاتابولیسم اسیدهای آمینه، تولید اسیدهای صفراوی و حذف هیدروژن پراکسید نقش دارند. بروز جهش در ژنهای PEX (مانند PEX1 که مسئول تقریباً 65 درصد موارد است، PEX6 و حداقل 13 ژن دیگر) موجب نقص یا کاهش شدید عملکرد پراکسیزومها میشود. این اختلال به تجمع مواد سمی در بدن و کمبود برخی محصولات ضروری منجر میگردد.
این بیماری در گروه اختلالات پراکسیزومی طبقهبندی میشود که شامل سه دسته اصلی است: اختلالات عمومی پراکسیزومی (مانند سندرم زلوگر، آدرنولوکودیستروفی نوزادی و بیماری رفسوم نوزادی)، اختلالات مربوط به حمل اسیدهای چرب زنجیره بلند (مانند آدرنولوکودیستروفی وابسته به کروموزوم X) و کندرودیسپلازی ریزوملیک پانکتات که با نقصهای چندگانه آنزیمی همراه است.

چرا این سندرم ایجاد می شود؟
سندرم زلوگر به دلیل اختلال در مسیرهای متابولیک مرتبط با پراکسیزومها شکل میگیرد و این نقصها تأثیری گسترده بر عملکرد بدن دارند. یکی از ویژگیهای این سندرم، تجمع اسیدهای چرب زنجیره بلند (VLCFAs) مانند C26 و C22 در پلاسما، فیبروبلاستها و آمنیوسیتها است که عواقب جدی به دنبال دارد. این تجمع میتواند غشاهای نورونی را تخریب کند، به دمیلیناسیون منجر شود، سمیت ناشی از هیدروژن پراکسید ایجاد کند و حتی سنتز هورمونهای استروئیدی را مختل کند. در سطح بافتی، مهمترین تغییرات شامل کیستهای کورتیکال در کلیه، فیبروز کبدی و ناهنجاریهای مغزی مشخص مانند دمیلیناسیون و پلیمیکروژیری سنتروسیلوین است. این بیماری تقریباً تمامی ارگانهای بدن را تحت تأثیر قرار میدهد، چرا که پراکسیزومها تقریباً در تمام سلولها، بهجز گلبولهای قرمز، وجود دارند. با این حال، تراکم بالای پراکسیزومها در کبد و کلیه تأثیر آسیبهای این بیماری در این دو اندام را برجستهتر نشان میدهد.
علائم بالینی
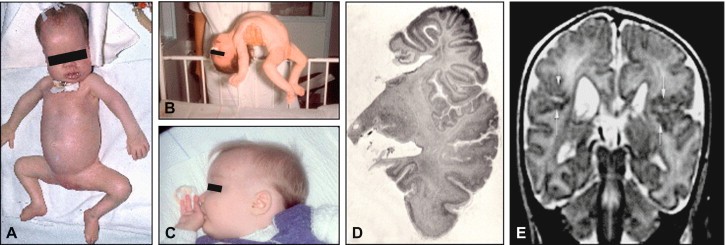
این سندرم، بهعنوان یکی از اختلالات نادر ژنتیکی، معمولاً در دوران نوزادی خود را نشان میدهد و ترکیبی از ناهنجاریهای مادرزادی و پیشرونده را به همراه دارد. این بیماری با مجموعهای از علائم واضح شناخته میشود که میتوان به ضعف شدید عضلانی (هیپوتونی)، کاهش حرکات خودبهخودی، گریه ضعیف، مشکلات تغذیهای، تشنجهای زودهنگام و شکلهای غیرعادی صورت مانند پیشانی بلند، فونتانل بزرگ، پل بینی پهن و سایر نشانههای دیسمورفیسم کرانیوفاشیال اشاره کرد.
همچنین، ناهنجاریهای چشمی مانند گلوکوم، کاتاراکت، مشکلات شبکیه، تیرگی قرنیه و آتروفی عصب بینایی نیز از دیگر علائم قابل توجه این بیماری هستند. از ویژگیهای دیگر این سندرم میتوان به اختلالات دندانی (نقص در مینای دندان)، هپاتومگالی (مشاهده شده در 80 درصد موارد) همراه با مشکلات آنزیمی کبد و کیستهای کلیوی که در 70 درصد بیماران دیده میشود اشاره کرد. مشکلات شنوایی حسی-عصبی، اختلالات جنسی مانند کریپتورکیدیسم و هایپوسپادیاس در مردان و کلیتورومگالی در زنان نیز ممکن است ظاهر شوند.
در دوره نوزادی، سه نشانه اصلی شامل هیپوتونی شدید، مشکلات تغذیه و تشنج غالب هستند. با رشد کودک، تأخیر شدید حرکتی-روانی، مشکلات رشد جسمانی و مسائل متابولیکی مانند سنگ کلیوی اگزالات کلسیم، نارسایی آدرنال و رگرسیون عصبی (بهدلیل لوکودیستروفی) آشکار میشوند. در مراحل بعدی زندگی، علائم خفیفتری مانند تأخیر رشد، آتاکسی مخچهای، نوروپاتی محیطی و دیگر علائم مرتبط با سیستم عصبی مشاهده میشود.
از سوی دیگر، ناهنجاریهای اسکلتی همچون کندرودیستروفی نقطهای (که عمدتاً در کشکک زانو و لگن دیده میشود) و کیستهای کلیوی زیر قشری نیز در میان بیماران شایع است. مطالعات تصویربرداری MRI مغزی نیز میتوانند به وجود ناهنجاریهایی همچون polymicrogyria در ناحیه پریسیلوین، گشاد شدن دوطرفه بطنها، تأخیر در میلیناسیون ماده سفید مغز یا دمیلیناسیون پیشرونده اشاره کنند.
تشخیص بالینی
تشخیص بالینی این سندرم با ارزیابی دقیق علائم آشکار آغاز میشود. پزشکان باید به ترکیبی از هیپوتونی، دیسمورفیسم صورت، هپاتومگالی، کیستهای کلیوی، تشنج، تأخیر رشدی، مشکلات شنوایی و بینایی، و اختلال عملکرد ارگانها توجه کنند. شدت و گستره علائم بر اساس سن بیمار در زمان بروز (نوزادی، کودکی، نوجوانی) بررسی میشود تا بتوان پیشآگهی را تعیین و آزمایشهای تکمیلی را برنامهریزی کرد.
تصویربرداری مانند سونوگرافی برای کیستهای کلیوی و MRI برای ارزیابی ناهنجاریهای مغزی نقش حمایتی دارند. علاوه بر این، غربالگری نوزادان امکان شناسایی موارد مرتبط را فراهم میکند؛ هرچند به صورت خاص برای زلوگر، غربالگری سطوحVLCFA بسیار حساس است.
تشخیص آزمایشگاهی
تشخیص آزمایشگاهی این سندرم بخش کلیدی فرآیند است و شامل آزمایشهای بیوشیمیایی و ژنتیکی میشود. مراحل اولیه این ارزیابی، اندازهگیری متابولیتهای مرتبط با فعالیت پراکسیزومی است که از طریق آزمایشهای زیر انجام میگیرد:
- سطوح VLCFA در پلاسما، فیبروبلاست یا آمنیوسیتها: افزایش C26:0، C26:1 و نسبتهای C24/C22 و C26/C22.
- کاهش سطوح پلاسمالوژنها در گلبولهای قرمز: شامل C16 و C18.
- افزایش اسید پیپکولیک در پلاسما: افزایش واسطههای اسیدهای صفراوی، فیتانیک و پریستانیک اسید.
در موارد خفیفتر که نتایج اولیه طبیعی هستند، بررسی فیبروبلاستهای کشتشده در دمای 40 درجه سانتیگراد گامی ضروری محسوب میشود. آزمایشهای عملکرد کبدی میزان افزایش آنزیمها یا بیلیروبین را نشان میدهند و ارزیابی عملکرد غدد فوق کلیوی نیز میتواند ناکارآمدی این سیستم را آشکار سازد. همچنین، آزمایش ژنتیکی از طریق پنل ژنتیکی PEX یا توالییابی کامل اگزوم یا ژنوم برای تشخیص جهشها (مانند جهش در PEX1 یا PEX6) انجام میشود. این روشها در تشخیص قطعی بیماری، شناسایی افراد حامل جهش ژنی و تشخیص پیش از تولد بسیار حائز اهمیت هستند. بر اساس الگوریتمهای پیشنهادی ACMG، توصیه میشود فرآیند تشخیص از آزمایشهای بیوشیمیایی آغاز شود و سپس با روشهای مولکولی مورد تأیید قرار گیرد، بهویژه هنگامیکه از تکنولوژی نسل بعدی توالییابی (NGS) استفاده شده باشد.
| روش تشخیصی | توضیح | کاربرد |
| آزمایش VLCFAs پلاسما | اندازهگیری افزایش C26:0 و نسبتها | غربالگری اولیه و حساس برای اختلالات پراکسیزومی |
| سطوح پلاسمالوژنها در گلبول های قرمز | کاهش C16/C18 پلاسمالوژنها | نشاندهنده نقص سنتز اترفسفولیپیدها |
| آزمایش اسید پیپکولیک | افزایش سطوح | حمایتکننده در تشخیص بیوشیمیایی |
| توالی یابی ژنتیکی ژن های PEX | شناسایی جهش در 13 ژن PEX | تشخیص قطعی و مشاوره ژنتیکی |
| آزمایش در فیبروبلاست ها | ارزیابی در دمای بالا برای موارد خفیف | وقتی آزمایشهای پلاسما طبیعی هستند |
| MRI مغز | شناسایی polymicrogyria و دمیلیناسیون | حمایت بالینی برای ارزیابی عصبی |
تشخیص افتراقی
معمولاً بر اساس علائم غالب بیمار انجام میشود:
- برای هیپوتونی در نوزادان، شرایطی همچون اختلالات کروموزومی (مانند سندرم داون و پرادر ویلی)، آتروفی نخاعی عضلانی، انسفالوپاتی هیپوکسیک-ایسکمیک و اختلالات پراکسیزومی (مانند کمبود آسیل کو آنزیم-ا اکسیداز یا پروتئین D-bifunctional) باید در نظر گرفته شوند.
- در شرایط کاهش شنوایی همراه با رتینیت پیگمنتوزا، سندرمهای آشر، کوکائین، آلپورت، واردنبرگ و رفسوم به عنوان دلایل احتمالی مطرح هستند.
- برای کاتاراکت دوطرفه، مواردی همچون سندرم Lowe، گالاکتوزمی، عفونتهای مادرزادی و کندرودیسپلازی ریزوملیک پانکتات بررسی میشوند.
- افتراق نارسایی آدرنوکورتیکال به عواملی نظیر خونریزی غده فوق کلیه، آدرنولکودیستروفی وابسته به کروموزوم X و آدرنالیت عفونی وابسته است.
تشخیص سندرم زلوگر با اندازهگیری افزایش VLCFAها (اسیدهای چرب زنجیره بلند) و بررسی جهشهای ژن PEX ممکن میشود.
پیش آگهی سندرم
پیشآگهی بیماری ضعیف بوده و به شدت علائم بستگی دارد. نوزادانی که مبتلا میشوند اغلب به دلیل نارسایی تنفسی، آپنه یا عفونتها در سال اول زندگی فوت میکنند. در کودکان موارد نارسایی پیشرونده کبدی کمی طولانیتر ادامه مییابد. در نوجوانان یا بزرگسالان جوان، بیماری ممکن است بقا را تا حدودی افزایش دهد اما معمولاً منجر به اسپاستیسیتی، نوروپاتی محیطی و افت شدید عصبی میشود. به طور کلی طول عمر به ندرت بیش از دو سال است و بیماران با مشکلاتی مثل خونریزی گوارشی، نارسایی کبدی، پنومونی، دیسترس تنفسی و عفونتهای مکرر مواجه خواهند شد. تنوع ژنتیکی و فنوتیپی نقش مهمی در پیشآگهی دارد، اما بیماری کماکان پیشرونده و کشنده باقی مانده است.
نحوه مدیریت
مدیریت سندرم زلوگر عمدتاً حمایتی است، چرا که درمان قطعی برای آن وجود ندارد:
- مصرف مکمل docosahexaenoic acid (DHA) برای جبران کمبود پلاسمایی پیشنهاد شده است، هرچند مطالعات مزایای واضحی برای بهبود عصبی یا بینایی نشان ندادهاند.
- روغن لورنزو به عنوان راهی برای کاهش VLCFAها استفاده میشود، اما تأثیر قابلتوجهی در توقف پیشرفت بیماری ندارد.
- اسید کولی (که FDA برای نقص سنتز اسیدهای صفراوی تأیید کرده است) ممکن است به جذب بهتر ویتامینهای محلول در چربی کمک کند.
- مراقبتهای حمایتی شامل استفاده از سمعک یا ایمپلنت حلزون شنوایی برای کاهش شنوایی، جراحی کاتاراکت و استفاده از عینک برای مشکلات بینایی، مدیریت تشنج با داروهای ضدتشنج، مکمل ویتامین K برای اصلاح مشکلات انعقادی، مصرف ویتامینهای محلول در چربی A، D و E، تجویز کورتیکواستروئید برای نارسایی آدرنال و استفاده از لوله گاستروستومی برای تغذیه میشود.
- همچنین توصیه میشود رژیم غذایی حاوی محدودیت برداشت اسید فیتانیک (مانند اجتناب از مصرف شیر گاو) رعایت شود.
Reference:
https://www.ncbi.nlm.nih.gov/books/NBK560676/
https://pubmed.ncbi.nlm.nih.gov/28409475/
https://www.sciencedirect.com/science/article/pii/S1098360021011539
https://www.orpha.net/en/disease/detail/912
https://www.physio-pedia.com/Zellweger_Syndrome