
مقدمه
سندرم رت یک اختلال ژنتیکی نادر است که عمدتاً دختران را تحت تأثیر قرار میدهد و باعث مشکلات شدید جسمی و ذهنی میشود. این بیماری با شیوع تقریبی ۱ در ۱۰,۰۰۰ تا ۱ در ۱۵,۰۰۰ تولد زنده، یکی از شایعترین علل ژنتیکی ناتوانی ذهنی در دختران است، پس از سندرم داون. پسران مبتلا به این بیماری نادر هستند و معمولاً علائم شدیدتری دارند که اغلب منجر به مرگ زودهنگام میشود. کودکان مبتلا به سندرم رت معمولاً در ۶ تا ۱۸ ماه اول زندگی به طور طبیعی رشد میکنند. پس از این دوره، آنها شروع به از دست دادن مهارتهای کسبشده مانند توانایی صحبت کردن، راه رفتن، یا استفاده هدفمند از دستها میکنند.

علائم کلیدی سندرم رت شامل موارد زیر است:
- از دست دادن مهارتهای دستی: کودکان توانایی استفاده هدفمند از دستها را از دست میدهند و حرکات تکراری مانند دستزدن یا مالیدن دستها ظاهر میشود.
- مشکلات گفتاری: توانایی صحبت کردن کاهش مییابد یا کاملاً از بین میرود.
- مشکلات حرکتی و هماهنگی: راه رفتن دشوار میشود و هماهنگی حرکتی کاهش مییابد.
- مشکلات تنفسی: تنفس نامنظم، مانند نگه داشتن نفس یا تنفس سریع، شایع است.
- تشنج: بسیاری از کودکان مبتلا تشنج را تجربه میکنند.
- ناتوانیهای ذهنی: مشکلات شناختی و یادگیری معمول هستند.
- کاهش رشد: رشد سر (میکروسفالی اکتسابی) و رشد کلی بدن کند میشود.
سندرم رت یک بیماری مادامالعمر است که تأثیر عمیقی بر زندگی فرد و خانوادهاش دارد. اگرچه درمانی برای این بیماری وجود ندارد، پیشرفتهای اخیر در درمانهای دارویی و ژندرمانی امیدهای جدیدی را برای بهبود کیفیت زندگی ایجاد کرده است.
تاریخچه و کشف سندرم رت
سندرم رت برای اولین بار در سال ۱۹۶۶ توسط دکتر آندریاس رت، یک متخصص اطفال اتریشی، توصیف شد. او متوجه شد که چندین دختر جوان در مطبش علائم مشابهی از جمله حرکات تکراری دست و از دست دادن مهارتهای زبانی را نشان میدهند. با این حال، تا سال ۱۹۸۳، زمانی که دکتر بنتسون هاگبرگ در سوئد مقالهای در مورد ۳۵ مورد منتشر کرد، سندرم رت به طور گستردهتری شناخته نشد.
در سال ۱۹۹۹، یک پیشرفت بزرگ رخ داد: دکتر هودا زوگبی و همکارانش در کالج پزشکی بیلور کشف کردند که جهش در ژن MECP2 روی کروموزوم X علت اصلی سندرم رت است. این کشف راه را برای تحقیقات بیشتر در مورد مکانیسمهای بیماری و توسعه درمانهای بالقوه هموار کرد.
از آن زمان، دانش در مورد سندرم رت به طور قابل توجهی گسترش یافته است، و سازمانهایی مانند بنیاد بینالمللی سندرم رت (IRSF) و اعتماد تحقیقاتی سندرم رت (RSRT) نقش مهمی در حمایت از تحقیقات و پشتیبانی از خانوادهها ایفا کردهاند.
علائم و مراحل سندرم رت
سندرم رت به طور کلی به چهار مرحله تقسیم میشود، هرچند که همه افراد لزوماً همه مراحل را تجربه نمیکنند یا به همان شدت:
- مرحله ۱: رکود اولیه (۶ تا ۱۸ ماه):
-
- رشد کمی کندتر از حد طبیعی.
- کاهش تماس چشمی و علاقه به اسباببازیها.
- تأخیر در نشستن یا خزیدن.
- مرحله ۲: تخریب سریع (۱ تا ۴ سال):
-
- از دست دادن سریع مهارتهای دستی و زبانی.
- بروز حرکات تکراری دست.
- مشکلات تنفسی، مانند نفس کشیدن سریع یا نگه داشتن نفس.
- گریه و بیقراری بیدلیل.
- تأخیر در رشد سر.
- مرحله ۳: فلات (۲ تا ۱۰ سال):
-
- بهبود نسبی در رفتار و ارتباطات.
- تشنجها ممکن است شروع شوند یا بدتر شوند.
- مشکلات حرکتی، مانند سفتی عضلانی و اسکولیوز.
- مرحله ۴: زوال حرکتی دیررس (پس از ۱۰ سال):
-
- کاهش تحرک، ضعف عضلانی، و سفتی.
- برخی افراد ممکن است توانایی راه رفتن را از دست بدهند.
- با این حال، شناخت و ارتباطات ممکن است پایدار بمانند یا بهبود یابند.
درک این مراحل میتواند به خانوادهها و مراقبتکنندگان کمک کند تا برای چالشهای پیش رو آماده شوند و مداخلات مناسب را برنامهریزی کنند.

پایه ژنتیکی سندرم رت
سندرم رت توسط جهشهایی در ژن MECP2 که روی کروموزوم X قرار دارد، ایجاد میشود. این ژن دستورالعملهایی برای تولید پروتئین MeCP2 ارائه میدهد که برای رشد و عملکرد طبیعی مغز ضروری است. پروتئین MeCP2 به تنظیم بیان ژنها کمک میکند و نقش مهمی در ارتباطات عصبی و انعطافپذیری سیناپسی ایفا میکند.
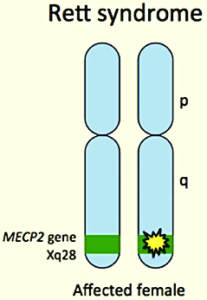
جهش در ژن MECP2 منجر به کمبود یا نقص عملکرد پروتئین MeCP2 میشود، که باعث بروز علائم سندرم رت میگردد. بیش از ۲۰۰ جهش مختلف در MECP2 شناسایی شده است که میتوانند شدت بیماری را تحت تأثیر قرار دهند.
از آنجا که ژن MECP2 روی کروموزوم X قرار دارد و زنان دو کروموزوم X دارند، یکی از این کروموزومها در هر سلول به طور تصادفی غیرفعال میشود (فرایندی به نام غیرفعالسازی کروموزوم X یا XCI). در دختران مبتلا به سندرم رت، کروموزوم X غیرفعال اغلب حاوی یک نسخه سالم از ژن MECP2 است، در حالی که کروموزوم فعال نسخه جهشیافته را دارد. این ترکیب باعث میشود شدت علائم در افراد مختلف متفاوت باشد.
انواع جهشهای MECP2
بیش از ۲۰۰ جهش مختلف در ژن MECP2 شناسایی شدهاند که شامل موارد زیر هستند:
- جهشهای نقطهای (Missense Mutations): شایعترین نوع، که منجر به تغییر یک آمینواسید در پروتئین MeCP2 میشود.
- جهشهای بیمعنی (Nonsense Mutations): باعث توقف زودرس سنتز پروتئین میشوند (مانند R168X، R255X).
- حذفها و درجها: حذف یا اضافه شدن نوکلئوتیدها که میتوانند ساختار پروتئین را مختل کنند.
- جهشهای اسپلایسینگ (Splice-Site Mutations): اختلال در پردازش RNA.
- حذفهای بزرگ: حذف بخشهایی از ژن که با MLPA قابل شناسایی است.
در مقابل، پسران که تنها یک کروموزوم X دارند، معمولاً در صورت داشتن جهش در MECP2 علائم شدیدتری را تجربه میکنند که اغلب منجر به مرگ در اوایل زندگی میشود. با این حال، برخی پسران با جهشهای خفیفتر یا موزاییسم (ترکیبی از سلولها با و بدون جهش) میتوانند زنده بمانند و علائم مشابه دختران مبتلا به سندرم رت را نشان دهند.
تشخیص بالینی سندرم رت
تشخیص بالینی معمولاً توسط متخصصین اطفال، نورولوژیستها، یا ژنتیستهای بالینی انجام میشود و باید با آزمایش ژنتیکی تأیید شود.
تشخیص سندرم رت در درجه اول بر اساس معیارهای بالینی انجام میشود که توسط گروههای بینالمللی مانند گروه کاری سندرم رت (RettSearch) و انجمن بینالمللی سندرم رت (IRSF) در سال ۲۰۱۰ تعریف شدهاند. این معیارها به چهار دسته اصلی و حمایتی تقسیم میشوند:
معیارهای اصلی تشخیصی (الزامی برای تشخیص کلاسیک سندرم رت)
- رگرسیون رشدی: دورهای از رشد طبیعی (معمولاً ۶ تا ۱۸ ماه اول زندگی) به دنبال از دست دادن مهارتهای کسبشده مانند زبان گفتاری و استفاده هدفمند از دستها.
- از دست دادن استفاده هدفمند از دستها: جایگزینی با حرکات تکراری استرئوتایپیک (مانند دستزدن، مالیدن دستها، یا فشار دادن).
- اختلال در راه رفتن: راه رفتن غیرطبیعی یا از دست دادن توانایی راه رفتن.
- کاهش رشد سر: میکروسفالی اکتسابی (کاهش رشد دور سر پس از تولد).
معیارهای حمایتی (برای حمایت از تشخیص)
- مشکلات تنفسی (هایپرونتیلاسیون، آپنه، یا تنفس نامنظم).
- تشنج.
- اسکولیوز.
- اسپاستیسیتی یا سفتی عضلانی.
- مشکلات گوارشی (مانند یبوست مزمن).
- کاهش پاسخ به درد.
- اختلالات خواب.
- الگوهای غیرطبیعی EEG.
معیارهای افتراقی
تشخیص سندرم رت باید با سایر اختلالات تکامل عصبی مانند اوتیسم، فلج مغزی، یا سایر اختلالات ژنتیکی مرتبط با کروموزوم X (مانند سندرم X شکننده) افتراق داده شود. مواردی که تشخیص سندرم رت را رد میکنند شامل تروما یا آسیب مغزی قبل از تولد، بیماریهای متابولیک پیشرونده، یا بیماریهای عصبی دیگر با علائم مشابه است.
تشخیص ژنتیکی سندرم رت
تشخیص ژنتیکی سندرم رت با شناسایی جهشهای ژن MECP2 انجام میشود. این ژن که روی کروموزوم X (Xq28) قرار دارد، پروتئینی را کد میکند که در تنظیم بیان ژنها و عملکرد سیناپسی مغز نقش دارد. بیش از ۹۵٪ موارد کلاسیک سندرم رت و حدود ۵۰-۷۰٪ موارد غیرکلاسیک (آتیپیکال) با جهشهای MECP2 مرتبط هستند.
روشهای آزمایش ژنتیکی
- توالییابی ژن MECP2 (Sanger Sequencing یاNext-Generation Sequencing ):
-
- توضیح: توالییابی ژن MECP2 برای شناسایی جهشهای نقطهای، حذفها، یا درجهای کوچک در مناطق کدکننده (اکسونها) و نواحی تنظیمکننده (اینترونها و پروموترها) انجام میشود.
- کاربرد: این روش استاندارد طلایی برای تشخیص سندرم رت است و معمولاً اولین گام در آزمایش ژنتیکی است.
- محدودیتها: ممکن است جهشهای بزرگ یا تغییرات ساختاری (مانند حذفهای بزرگ) را شناسایی نکند.
- آزمایش MLPA (Multiplex Ligation-dependent Probe Amplification):
-
- توضیح: MLPA برای تشخیص حذفها یا مضاعفشدگیهای بزرگ در ژن MECP2 استفاده میشود که ممکن است با توالییابی شناسایی نشوند.
- کاربرد: در مواردی که توالییابی نتیجه منفی دارد، اما علائم بالینی قویاً به سندرم رت اشاره دارند.
- محدودیتها: حساسیت محدود به تغییرات ساختاری خاص.
- پنلهای ژنتیکی NGS:
-
- توضیح: پنلهایNGS میتوانند چندین ژن مرتبط با اختلالات تکامل عصبی (مانند MECP2، CDKL5، FOXG1) را به طور همزمان بررسی کنند.
- کاربرد: در مواردی که علائم غیرکلاسیک هستند یا تشخیص افتراقی با سایر اختلالات مورد نیاز است.
- مزایا: افزایش احتمال شناسایی ژنهای دیگر مرتبط با سندرم رت آتیپیکال (مانند CDKL5 و FOXG1)
- Whole-Exome Sequencing (WES):
-
- توضیح: توالییابی کل اگزوم برای شناسایی جهشهای نادر در ژنهای کمتر شناختهشدهای که ممکن است علائم مشابه سندرم رت ایجاد کنند.
- کاربرد: در مواردی که آزمایشهای MECP2 منفی هستند، اما علائم بالینی قویاً به یک اختلال ژنتیکی اشاره دارند.
- محدودیتها: هزینه بالا و پیچیدگی در تفسیر نتایج.
- Whole-Genome Sequencing (WGS):
-
- توضیح: بررسی کل ژنوم برای شناسایی جهشهای غیرکدکننده یا تغییرات ساختاری پیچیده.
- کاربرد: در موارد تحقیقاتی یا زمانی که سایر روشها نتیجهای نداشته باشند.
- محدودیتها: هزینه بسیار بالا و نیاز به تحلیل بیوانفورماتیک پیشرفته.
پروتکل پیشنهادی برای آزمایش ژنتیکی
- گام اول: توالییابی ژن MECP2 (NGS یا Sanger) برای شناسایی جهشهای نقطهای و تغییرات کوچک.
- گام دوم: در صورت منفی بودن نتیجه، انجام آزمایش MLPA برای تشخیص حذفها یا دوپلیکاسیونهای بزرگ.
- گام سوم: اگر نتایج همچنان منفی باشند و علائم بالینی قوی باشند، استفاده از پانل NGS یا WES برای بررسی ژنهای دیگر مانند CDKL5 و FOXG1.
- گام چهارم: در موارد پیچیده، WGS برای شناسایی تغییرات غیرمعمول یا نادر.
موارد خاص در تشخیص ژنتیکی
- سندرم رت آتیپیکال: در این موارد، علائم ممکن است خفیفتر یا متفاوت باشند و جهشهای MECP2 در حدود ۵۰-۷۰٪ موارد یافت میشوند. ژنهای دیگر مانند CDKL5 (مرتبط با سندرم رت واریانت با تشنج زودرس) و FOXG1 (مرتبط با سندرم رت مادرزادی) باید بررسی شوند.
- موارد منفی MECP2: حدود ۵٪ از موارد کلاسیک و ۳۰-۵۰٪ از موارد آتیپیکال هیچ جهش قابل شناسایی در MECP2 ندارند. این ممکن است به دلیل جهشهای غیرکدکننده، تغییرات اپیژنتیکی، یا ژنهای دیگر باشد.
- موارد پسران: پسران با جهش MECP2 معمولاً علائم شدیدتری دارند و ممکن است در اوایل کودکی فوت کنند، اما موارد خفیفتر (مانند موزاییسم یا جهشهای جزئی) نیز گزارش شدهاند.
ابزارهای تشخیصی مکمل
- الکتروانسفالوگرام (EEG): برای ارزیابی فعالیت تشنجی و الگوهای غیرطبیعی مغزی.
- تصویربرداری مغزی (MRI/CT): برای رد سایر علل نورولوژیک مانند ضایعات ساختاری.
- آزمایشهای متابولیک: برای افتراق با بیماریهای متابولیک پیشرونده.
مشاوره ژنتیکی برای سندرم رت
مشاوره ژنتیکی بخش ضروری مدیریت سندرم رت است و باید توسط متخصصین ژنتیک بالینی یا مشاوران ژنتیک آموزشدیده ارائه شود. هدف مشاوره ژنتیکی، ارائه اطلاعات دقیق، حمایت عاطفی، و راهنمایی برای تصمیمگیریهای آگاهانه به خانوادهها است.
مراحل مشاوره ژنتیکی
- جمعآوری تاریخچه پزشکی و خانوادگی:
-
- تاریخچه بیمار: بررسی دقیق علائم، مراحل رشدی، و نتایج آزمایشهای بالینی و ژنتیکی.
- شجرهنامه خانوادگی: تهیه شجرهنامه سهنسلی برای شناسایی الگوهای ارثی یا حاملان احتمالی.
- سابقه بیماریهای مرتبط: بررسی سایر اختلالات تکامل عصبی یا ژنتیکی در خانواده.
- تفسیر نتایج آزمایش ژنتیکی:
-
- جهشهای شناساییشده: توضیح نوع جهش (مانند جهش نقطهای یا حذف)، تأثیر آن بر پروتئین MeCP2، و ارتباط با شدت بیماری.
- موارد منفی: در صورت عدم شناسایی جهش در MECP2، بحث در مورد احتمال ژنهای دیگر (مانند CDKL5، FOXG1) یا محدودیتهای آزمایش.
- موزاییسم: در مواردی که موزاییسم (ترکیب سلولهای جهشیافته و سالم) شناسایی شود، توضیح تأثیر آن بر شدت بیماری و پیشآگهی.
- ارزیابی ریسک ارثی:
-
- جهشهای de novo: بیش از ۹۹٪ موارد سندرم رت ناشی از جهشهای جدید (de novo) هستند و ریسک تکرار در فرزندان بعدی والدین بسیار پایین است .
- جهشهای ارثی: در موارد نادر، جهش ممکن است از مادر حامل (اغلب بدون علامت به دلیل غیرفعالسازی کروموزوم X) به ارث برسد. آزمایش ژنتیکی مادر برای شناسایی وضعیت حامل ضروری است.
- ریسک در پسران: اگر مادر حامل باشد، احتمال انتقال جهش به پسران ۵۰٪ است، که معمولاً منجر به علائم شدید یا مرگ زودهنگام میشود.
- موزاییسم گونادال: در برخی موارد، والدین ممکن است موزاییسم گونادال داشته باشند (جهش فقط در سلولهای جنسی)، که ریسک تکرار را افزایش میدهد.
- بحث در مورد پیشآگهی و مدیریت:
-
- پیشآگهی: شدت علائم بسته به نوع جهش و الگوی غیرفعالسازی کروموزوم X متفاوت است. به عنوان مثال، جهشهای R168X و R255X معمولاً با علائم شدیدتر مرتبط هستند.
- مدیریت: معرفی درمانهای موجود (مانند تروفینتید، فیزیوتراپی، و کاردرمانی) و بحث در مورد آزمایشهای بالینی ژندرمانی (مانند TSHA-102 و NGN-401).
- منابع حمایتی: ارجاع به سازمانهایی مانند IRSF و RSRT برای پشتیبانی و آموزش.
- گزینههای تولیدمثلی:
-
- تشخیص پیش از لانهگزینی (PGD): برای خانوادههایی با جهش ارثی شناختهشده، PGD میتواند برای انتخاب جنینهای بدون جهش استفاده شود.
- تشخیص پیش از تولد: آزمایش ژنتیکی جنین از طریق نمونهگیری از پرزهای کوریونی (CVS) یا آمنیوسنتز برای شناسایی جهش MECP2.
- مشاوره در مورد ریسک تکرار: توضیح ریسک پایین در جهشهای de novo و ریسک بالاتر در موارد ارثی یا موزاییسم گونادال.
- حمایت روانی-اجتماعی:
-
- حمایت عاطفی: کمک به خانوادهها برای کنار آمدن با تشخیص و تأثیر آن بر زندگی.
- ارجاع به گروههای حمایتی: معرفی به سازمانهای محلی و بینالمللی مانند IRSF و RSRT.
- آموزش خانواده: ارائه اطلاعات در مورد مراقبتهای روزمره، مدیریت تشنج، و بهبود کیفیت زندگی.
ملاحظات خاص در مشاوره ژنتیکی
- تنوع در شدت بیماری: شدت علائم به الگوی غیرفعالسازی کروموزوم X (XCI skewing) بستگی دارد. در برخی موارد، غیرفعالسازی ترجیحی کروموزوم X جهشیافته میتواند علائم را خفیفتر کند.
- مشاوره برای پسران مبتلا: در پسران، سندرم رت نادر است و معمولاً با پیشآگهی ضعیف همراه است. مشاوره باید شامل بحث در مورد احتمال مرگ زودهنگام یا علائم شدید باشد.
- موارد آتیپیکال: در مواردی که جهش در MECP2 یافت نمیشود، باید احتمال اختلالات مرتبط (مانند سندرم CDKL5 یا FOXG1) بررسی شود.
- اخلاقیات: بحث در مورد تصمیمگیریهای تولیدمثلی و حریم خصوصی ژنتیکی باید با حساسیت انجام شود.
پیشرفتهای اخیر در تشخیص سندرم رت
پیشرفتهای تشخیصی
- آزمایشهای اپیژنتیکی: بررسی الگوهای غیرفعالسازی کروموزوم X (XCI) با استفاده از تکنیکهایی مانند PCR مبتنی بر متیلاسیون برای درک بهتر شدت بیماری.
- بیومارکرها: تحقیقات در حال بررسی بیومارکرهای مغزی (مانند سطح پروتئین MeCP2 یا BDNF) برای کمک به تشخیص و ارزیابی پاسخ به درمان هستند.
درمان سندرم رت
تا سال ۲۰۲۳، درمانهای سندرم رت بر مدیریت علائم متمرکز بود. این درمانها شامل موارد زیر هستند:
- فیزیوتراپی: برای بهبود تحرک، تقویت عضلات، و جلوگیری از سفتی مفاصل.
- کاردرمانی: برای تقویت مهارتهای استفاده از دست، فعالیتهای روزمره، و ارتباطات.
- گفتاردرمانی: برای حمایت از ارتباطات غیرکلامی، مانند استفاده از تصاویر یا دستگاههای ارتباطی.
- داروها: برای کنترل تشنجها (مانند داروهای ضد تشنج)، مشکلات تنفسی (مانند داروهای ضد اضطراب)، یا اضطراب و افسردگی.
- حمایت تغذیهای: برای مدیریت مشکلات تغذیه و رشد.
در مارس ۲۰۲۳، سازمان غذا و داروی آمریکا (FDA) داروی تروفینتید (با نام تجاری Daybue) را به عنوان اولین درمان خاص برای سندرم رت تأیید کرد. تروفینتید یک آنالوگ مصنوعی از گلیسین-پرولین-گلوتامات، بخشی از پروتئین فاکتور رشد شبه انسولین ۱ (IGF-1)، است. این دارو با کاهش التهاب مغز، جلوگیری از فعالیت بیش از حد برخی سلولها، و افزایش سطح پروتئین IGF-1 عمل میکند.
مطالعات بالینی، از جمله مطالعه فاز سوم LAVENDER، نشان داد که تروفینتید بهبود قابلتوجهی در علائم اصلی سندرم رت ایجاد میکند، همانطور که در پرسشنامه رفتار سندرم رت (RSBQ) و مقیاس جهانی بالینی بهبود (CGI-I) اندازهگیری شد. بیماران تحت درمان با تروفینتید بهبودهایی در ارتباطات، تعاملات اجتماعی، و رفتارهای کلی نشان دادند. عوارض جانبی شایع شامل اسهال و استفراغ است.
تروفینتید یک پیشرفت بزرگ است، اما درمان قطعی نیست و نیاز به درمانهای مؤثرتر همچنان وجود دارد.

درمانهای مکمل و جایگزین
علاوه بر درمانهای پزشکی، برخی خانوادهها از درمانهای مکمل و جایگزین برای بهبود کیفیت زندگی فرزندانشان استفاده میکنند. این درمانها ممکن است شامل موارد زیر باشند:
- موسیقیدرمانی: برای تقویت ارتباطات، کاهش اضطراب، و بهبود خلق و خو.
- آبدرمانی: برای بهبود تحرک و تقویت عضلات در محیط کمفشار.
- رژیمهای غذایی خاص: برخی خانوادهها رژیمهای غذایی بدون گلوتن یا کازئین را امتحان میکنند، اگرچه شواهد علمی محدودی برای حمایت از این رویکردها وجود دارد.
- مکملهای غذایی: مانند اسیدهای چرب امگا-۳، که ممکن است به بهبود عملکرد مغز کمک کنند.
- یوگا و مدیتیشن: برای کاهش استرس و بهبود تمرکز.
مهم است که خانوادهها قبل از شروع هرگونه درمان مکمل با پزشک مشورت کنند، زیرا برخی از این درمانها ممکن است با داروهای دیگر تداخل داشته باشند یا عوارض جانبی داشته باشند.

تأثیر بر خانوادهها و مراقبتکنندگان
سندرم رت نه تنها بر فرد مبتلا، بلکه بر کل خانواده تأثیر میگذارد. مراقبت از یک کودک با نیازهای ویژه میتواند چالشبرانگیز باشد و خانوادهها ممکن است با استرس عاطفی، مالی، و فیزیکی روبرو شوند.
برخی از چالشهای رایج عبارتند از:
- مراقبت ۲۴ ساعته: بسیاری از کودکان مبتلا به سندرم رت نیاز به نظارت مداوم دارند، به ویژه اگر تشنج یا مشکلات تنفسی داشته باشند.
- هزینههای پزشکی: درمانها، تجهیزات، و مراقبتهای ویژه میتوانند گران باشند.
- تأثیر بر خواهر و برادرها: خواهر و برادرها ممکن است احساس کنند توجه کمتری دریافت میکنند یا مسئولیتهای بیشتری بر عهده دارند.
- انزوای اجتماعی: خانوادهها ممکن است به دلیل محدودیتهای حرکتی یا نیازهای مراقبتی، کمتر در فعالیتهای اجتماعی شرکت کنند.
با این حال، منابع حمایتی بسیاری برای خانوادهها وجود دارد، از جمله:
- گروههای حمایتی: آنلاین یا حضوری، که خانوادهها میتوانند تجربیات خود را به اشتراک بگذارند و از یکدیگر حمایت کنند.
- خدمات مشاوره: برای کمک به مدیریت استرس و اضطراب.
- برنامههای آموزشی: برای آموزش مهارتهای مراقبتی و مدیریت رفتار.
- کمکهای مالی: از طریق سازمانهای خیریه یا برنامههای دولتی.
سازمانهایی مانند بنیاد بینالمللی سندرم رت (IRSF) و اعتماد تحقیقاتی سندرم رت (RSRT) منابع، آموزش، و پشتیبانی را برای خانوادهها ارائه میدهند.
رویکردهای نویدبخش ژندرمانی
ژندرمانی به عنوان یک روش نوآورانه برای درمان اختلالات ژنتیکی مانند سندرم رت در حال بررسی است. این روش با هدف اصلاح یا جبران ژن معیوب، در این مورد ژن MECP2، عمل میکند.
مطالعه دانشگاه UC Davis
تحقیقات اخیر به رهبری سانچیتا باتناگار در دانشگاه UC Davis Health رویکرد جدیدی را برای درمان سندرم رت ارائه داده است. این مطالعه که در ژورنال Nature Communications منتشر شد، بر فعالسازی مجدد نسخه سالم ژن MECP2 که در کروموزوم X غیرفعال قرار دارد، تمرکز دارد.
محققان دریافتند که یک مولکول کوچک RNA به نام miR-106a نقش مهمی در غیرفعالسازی کروموزوم X و خاموش کردن ژن MECP2 دارد. آنها یک بردار ژندرمانی طراحی کردند که یک مولکول DNA به نام “اسفنج” را تحویل میدهد که miR-106a را جذب کرده و اثر خاموشکننده آن را کاهش میدهد. این روش به فعالسازی مجدد ژن سالم MECP2 و تولید پروتئین MeCP2 کمک میکند.
در آزمایش روی مدل موشهای مبتلا به سندرم رت، این درمان نتایج چشمگیری داشت:
- افزایش طول عمر: موشهای درمانشده عمر طولانیتری داشتند (میانه حدود ۲۹. ۵۷ هفته در مقابل ۱۲. ۱۴ هفته برای گروه کنترل).
- بهبود حرکت و شناخت: موشها عملکرد حرکتی و شناختی بهتری نشان دادند.
- بهبود تنفس: الگوهای تنفسی نامنظم بهبود یافت.
- افزایش سطح MeCP2: سطح پروتئین MeCP2 به حدود ۳۲ درصد سطح طبیعی رسید.
این رویکرد به دلیل استفاده از ژن سالم موجود در بدن بیمار، پتانسیل ارائه یک درمان طبیعیتر و مؤثرتر را دارد. با این حال، پیش از آزمایشهای انسانی، مطالعات ایمنی بیشتری مورد نیاز است.

آزمایشهای بالینی در حال انجام
علاوه بر مطالعه UC Davis، شرکتهای بیوتکنولوژی در حال آزمایش ژندرمانیهای دیگر برای سندرم رت هستند:
- Taysha Gene Therapies (TSHA-102): این شرکت در حال انجام آزمایشهای فاز ۱/۲ برای TSHA-102 است که یک نسخه کوتاهشده (“مینیژن”) از ژن MECP2 را با استفاده از ویروس وابسته به آدنو (AAV) تحویل میدهد. این درمان شامل فناوری miRARE است که بیان ژن را تنظیم میکند تا از تولید بیش از حد پروتئین MeCP2 جلوگیری کند، زیرا این امر میتواند مضر باشد. دادههای اولیه از آزمایشهای بالینی در بزرگسالان و کودکان بهبودهایی در مهارتهای حرکتی، ارتباطات، و فعالیت تشنجی نشان داده است.
- Neurogene (NGN-401): این شرکت نیز آزمایش فاز ۱/۲ را برای ژندرمانی NGN-401 در کودکان مبتلا به سندرم رت انجام میدهد. دادههای اولیه نشاندهنده بهبود در پرسشنامه رفتار سندرم رت (RSBQ)، خواب، گوارش، و نقاط عطف رشدی است. با این حال، یک عارضه جانبی جدی در دوز بالا گزارش شده که نیاز به بررسیهای ایمنی بیشتر را نشان میدهد.
جدول زیر خلاصهای از این رویکردها ارائه میدهد:
| رویکرد | سازمان/شرکت | روش | وضعیت | نتایج اولیه |
| فعالسازی ژن سالم MECP2 | UC Davis Health | استفاده از اسفنج miR-106a برای کاهش خاموشی کروموزوم X | مطالعه پیشبالینی (موش) | افزایش طول عمر، بهبود حرکت، شناخت، و تنفس در موشها |
| TSHA-102 | Taysha Gene Therapies | تحویل مینیژن MECP2 با فناوری miRARE | آزمایش فاز ۱/۲ (بزرگسالان و کودکان) | بهبود در مهارتهای حرکتی، ارتباطات، و کاهش فعالیت تشنجی |
| NGN-401 | Neurogene | ژندرمانی برای تحویل ژن MECP2 | آزمایش فاز ۱/۲ (کودکان) | بهبود در رفتار، خواب، گوارش، و نقاط عطف رشدی؛ عارضه جانبی در دوز بالا گزارش شد |
تحقیقات آینده و مسیرهای پژوهشی
تحقیقات در مورد سندرم رت به سرعت در حال پیشرفت است و چندین مسیر پژوهشی امیدوارکننده در حال بررسی هستند:
- ویرایش ژن: فناوریهایی مانند CRISPR-Cas9 میتوانند برای اصلاح جهشهای ژن MECP2 در سلولها استفاده شوند. اگرچه این روش هنوز در مراحل اولیه است، پتانسیل ارائه یک درمان قطعی را دارد.
- سلولهای بنیادی: تحقیقات در حال بررسی استفاده از سلولهای بنیادی برای جایگزینی یا ترمیم سلولهای آسیبدیده در مغز است.
- درمانهای دارویی: داروهای جدیدی که بر مسیرهای مغزی خاص تأثیر میگذارند، مانند مهارکنندههای HDAC یا فعالکنندههای BDNF، در حال توسعه هستند.
- مدلهای حیوانی پیشرفته: توسعه مدلهای حیوانی بهتر، مانند مدلهای میمون، میتواند به درک بهتر بیماری و آزمایش درمانهای جدید کمک کند.
- پزشکی شخصی: با توجه به تنوع جهشهای MECP2، درمانهای شخصیسازیشده بر اساس ژنوتیپ فرد میتوانند مؤثرتر باشند.
حمایت مداوم از تحقیقات و مشارکت در آزمایشهای بالینی برای تبدیل این پیشرفتهای علمی به درمانهای مؤثر بسیار مهم است.
پرسشهای متداول (FAQ)
- آیا سندرم رت قابل پیشگیری است؟
-
- خیر، سندرم رت یک اختلال ژنتیکی است و در حال حاضر راهی برای پیشگیری از آن وجود ندارد. با این حال، مشاوره ژنتیکی میتواند به خانوادهها در درک خطرات کمک کند.
- آیا درمانی برای سندرم رت وجود دارد؟
-
- در حال حاضر، درمانی وجود ندارد، اما درمانهای دارویی و ژندرمانی در حال توسعه هستند و میتوانند علائم را بهبود بخشند.
- آیا سندرم رت ارثی است؟
-
- در بیشتر موارد، سندرم رت به دلیل جهشهای جدید در ژن MECP2 رخ میدهد و ارثی نیست. با این حال، در موارد نادر، جهش میتواند از والدین به ارث برسد.
- آیا افراد مبتلا به سندرم رت میتوانند به مدرسه بروند؟
-
- بله، بسیاری از کودکان مبتلا به سندرم رت میتوانند در برنامههای آموزشی ویژه شرکت کنند و از آموزشهای مناسب بهرهمند شوند.
- آیا سندرم رت بر طول عمر تأثیر میگذارد؟
-
- بله، سندرم رت میتواند بر طول عمر تأثیر بگذارد، به ویژه در موارد شدید یا در پسران. با این حال، بسیاری از افراد مبتلا با مراقبتهای مناسب میتوانند تا بزرگسالی زندگی کنند.
واژهنامه
- ژن: بخشی از DNA که دستورالعملهایی برای ساخت پروتئینها ارائه میدهد.
- جهش: تغییر در توالی DNA که میتواند منجر به بیماری شود.
- کروموزوم X: یکی از دو کروموزوم جنسی که در زنان دو نسخه و در مردان یک نسخه دارد.
- غیرفعالسازی کروموزوم X (XCI): فرایندی که در آن یکی از دو کروموزوم X در زنان به طور تصادفی غیرفعال میشود.
- پروتئین MeCP2: پروتئینی که توسط ژن MECP2 تولید میشود و برای عملکرد طبیعی مغز ضروری است.
- ژندرمانی: روشی برای درمان بیماریها با اصلاح یا جبران ژنهای معیوب.
میکروسفالی: رشد ناکافی سر و مغز.
Reference:
International Rett Syndrome Foundation (IRSF): https: //www.rettsyndrome.org
Rett Syndrome Research Trust (RSRT): https: //reverserett.org
ClinicalTrials.gov: https: //clinicaltrials.gov
Rett Syndrome Europe: https: //www.rettsyndrome.eu
National Institute of Neurological Disorders and Stroke (NINDS): https: //www.ninds.nih.gov
Rett Girl: https: //www.rettgirl.org
Girl Power 2 Cure (GP2C): https: //www.girlpower2cure.org
Rett Syndrome Angels: https: //www.rettsyndromeangels.org
Northwest Rett Syndrome Association (NWRSA): https: //www.nwrettsyndrome.org
Nature Communications Study (UC Davis): https: //www.nature.com/articles/s41467-025-61092-7