مقدمه
سندرم مارفان (Marfan Syndrome, MFS) یک اختلال ژنتیکی اتوزومال غالب است که بافت همبند بدن را تحت تأثیر قرار میدهد. این بیماری با شیوع تقریبی 1 در 3000 تا 5000 نفر در سراسر جهان، ناشی از جهش در ژن FBN1 واقع در کروموزوم 15q21.1 است که پروتئین فیبریلین-1 را کد میکند. فیبریلین-1 جزء اصلی میکروفیبریلها در ماتریکس خارج سلولی است و نقش مهمی در استحکام و انعطافپذیری بافتهای همبند، از جمله آئورت، چشمها و سیستم اسکلتی دارد. این بیماری میتواند طیف وسیعی از علائم را ایجاد کند که شدت آنها در افراد مختلف متفاوت است، از علائم خفیف تا موارد شدید مانند سندرم مارفان نوزادی که میتواند تهدیدکننده حیات باشد.
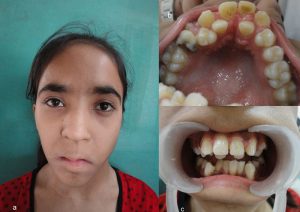
در این بیماران وضعیت دندانها هم دچار مشکل شده و مشکلات ارتودنسی ایجاد میشود.
علائم بالینی
سندرم مارفان با درگیری چندین سیستم بدن مشخص میشود. علائم اصلی شامل موارد زیر است:
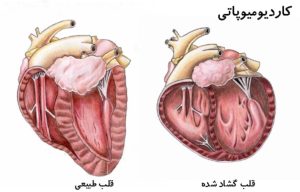
سیستم قلبی-عروقی
- اتساع ریشه آئورت و دیسکسیون آئورت: شایعترین و جدیترین عارضه که میتواند منجر به پارگی آئورت شود.
- پرولاپس دریچه میترال: شل شدن دریچه میترال که ممکن است باعث نارسایی قلبی شود.
- نارسایی آئورت: به دلیل ضعف بافت همبند در ریشه آئورت.
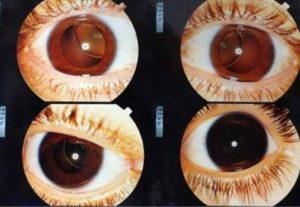
سیستم چشمی
- اکتوپیا لنتیس (دررفتگی عدسی): در 60-30% موارد، معمولاً قبل از 10 سالگی ظاهر میشود.
- نزدیکبینی (میوپیا): شایع و ممکن است شدید باشد.
- جدا شدن شبکیه: خطر افزایشیافتهای برای این عارضه وجود دارد.
- گلوکوم و کاتاراکت: در برخی موارد دیده میشود.
سیستم اسکلتی
- قد بلند و اندامهای کشیده: نسبت بازو به ارتفاع بیش از 1.05.
- آراکنوداکتیلی (انگشتان دراز): علامت مچ دست و شست مثبت.
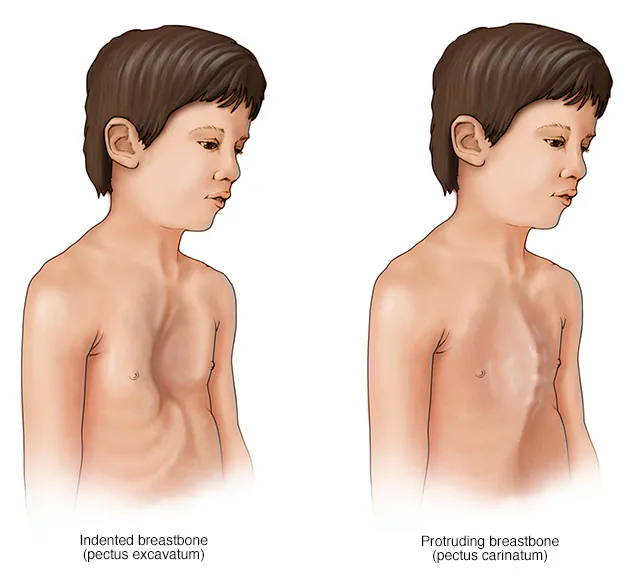
- پکتوس اکساواتوم یا کاریناتوم: فرورفتگی یا برآمدگی قفسه سینه.
- اسکولیوز: انحنای ستون فقرات در 18% موارد شدید است.
- پاهای صاف و کاهش امتداد آرنج: شایع در کودکان.
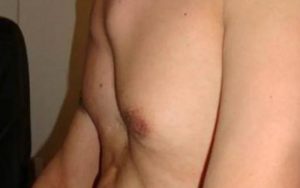
در سندروم مارفان ممکن است که استخوان سینه در قفسه سینه فرو رود.
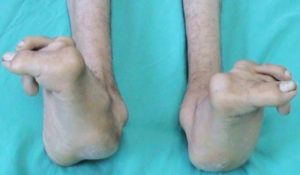
درصورتیکه بیماری مدیریت نشود، ممکن است استخوانها بدشکل شوند.
سایر سیستمها
- پنوموتوراکس خودبهخودی: به دلیل ضعف بافت ریه.
- استریا (خطوط پوستی): به دلیل کشیدگی پوست.
- فتق (اینگوینال یا شکمی): به دلیل ضعف دیواره شکم.
- درد کمر و مفاصل: ناشی از شلی مفاصل.
جدول 1: مروری بر علایم بالینی سندروم مارفان
| دستهبندی | علائم سندروم مارفان |
| قلب و عروق | اتساع آئورت، نارسایی دریچههای قلبی، خطر پارگی آئورت |
| چشمی | نزدیکبینی شدید، دررفتگی عدسی چشم، خطر آب مروارید و گلوکوم زودرس |
| اسکلتی | قد بلند غیرطبیعی، دستها و انگشتان بلند، ستون فقرات خمیده (اسکولیوز)، مفاصل بیش از حد انعطافپذیر |
| ریوی | احتمال بالای پنوموتوراکس (سوراخ شدن ریه)، مشکلات تنفسی |
| پوستی | ایجاد ترکهای پوستی بدون تغییر وزن زیاد |
انواع سندرم مارفان
سندرم مارفان به طور کلی به عنوان یک بیماری واحد با شدت متغیر شناخته میشود، اما میتوان آن را به دو دسته اصلی تقسیم کرد:
- سندرم مارفان کلاسیک: با طیف وسیعی از علائم خفیف تا متوسط که در بزرگسالی ظاهر میشوند.
- سندرم مارفان نوزادی: یک فرم شدید که معمولاً در بدو تولد یا اوایل کودکی تشخیص داده میشود و با عوارض قلبی-عروقی شدید و مرگ زودهنگام همراه است. این نوع اغلب با جهشهای خاص در اگزونهای 32-24 ژن FBN1 مرتبط است.
اختلالات مرتبط مانند سندرم لویز-دیتز (Loeys-Dietz Syndrome) یا سندرم اهلرز-دانلوس (Ehlers-Danlos Syndrome) ممکن است علائم مشابهی داشته باشند، اما از نظر ژنتیکی و بالینی متمایز هستند.
پاتوفیزیولوژی
سندرم مارفان به دلیل جهش در ژن FBN1 ایجاد میشود که منجر به تولید فیبریلین-1 معیوب میشود. فیبریلین-1 جزء اصلی میکروفیبریلها در ماتریکس خارج سلولی است که به استحکام بافتهای همبند کمک میکند و در تنظیم سیگنالینگ فاکتور رشد تبدیلکننده بتا (TGF-beta) نقش دارد. جهشهای FBN1 باعث موارد زیر می شود:
- ضعف میکروفیبریلها: کاهش استحکام بافتهای همبند، بهویژه در آئورت، چشمها و استخوانها.
- افزایش فعالیت TGF-beta: منجر به التهاب، فیبروز و بازسازی عروقی غیرطبیعی.
- تخریب دیواره آئورت: به دلیل تجمع موکوپلیساکاریدها و از دست دادن سلولهای عضلانی صاف، که به اتساع و دیسکسیون آئورت منجر میشود.
این تغییرات پاتوفیزیولوژیک مسئول علائم بالینی متنوع سندرم مارفان هستند.
روشهای تشخیص
تشخیص سندرم مارفان بر اساس معیارهای بالینی و ژنتیکی انجام میشود:
- معیارهای Ghent nosology (2010): این معیارها وزن بیشتری به تظاهرات قلبی-عروقی و اکتوپیا لنتیس میدهند. در غیاب سابقه خانوادگی، وجود این دو ویژگی برای تشخیص کافی است. در غیر این صورت، امتیاز سیستمیک (بر اساس ویژگیهای اسکلتی، چشمی و غیره) یا جهش FBN1 لازم است.
- تصویربرداری:
- اکوکاردیوگرافی: برای ارزیابی اندازه ریشه آئورت و عملکرد دریچهها.
- CT/MRI قفسه سینه و پایین ستون فقرات: برای شناسایی اتساع آئورت یا دکتازی دورال.
- معاینه چشم با اسلیتلمپ: برای تشخیص اکتوپیا لنتیس و گلوکوم.
- آزمایش ژنتیکی: شناسایی جهش در ژن FBN1 با استفاده از توالییابی کل اگزوم (WES) یا توالییابی Sanger. در ایران، مطالعاتی مانند مورد گزارششده در بیرجند از WES برای شناسایی جهشهای جدید استفاده کردهاند.
علل ژنتیکی
سندرم مارفان به دلیل جهش در ژن FBN1 ایجاد میشود که در جایگاه کروموزومی 15q21.1 قرار دارد. این ژن شامل 68 اگزون است و بیش از 90% بیماران مبتلا به سندرم مارفان دارای جهشهای قابل شناسایی در این ژن هستند. انواع جهشها شامل:
- جهشهای missense: تغییر یک آمینواسید، شایعترین نوع.
- جهشهای nonsense: ایجاد کدون توقف زودرس.
- جهشهای frameshift: درج یا حذف نوکلئوتیدها که باعث تغییر چارچوب خواندن میشود.
- جهشهای splicing: اختلال در پردازش RNA.
حدود 25% موارد به دلیل جهشهای de novo (غیرارثی و جدید) هستند. در کمتر از 10% بیماران با فنوتیپ کلاسیک مارفان، جهش FBN1 شناسایی نمیشود، که ممکن است به دلیل حذف کامل آلل یا تغییرات در تنظیم ژن باشد. جهشهای خاص در اگزونهای 32-24 با فنوتیپهای شدیدتر، مانند سندرم مارفان نوزادی، مرتبط هستند. به عنوان مثال، یک مطالعه در ایران یک جهش جدید missense (c.2179T>C/p.C727R) را در اگزون 19 شناسایی کرد که با فنوتیپ شدید همراه بود.
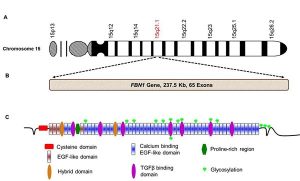
همبستگی ژنوتیپ-فنوتیپ
تحقیقات نشان دادهاند که نوع و محل جهش در FBN1 میتواند بر شدت بیماری تأثیر بگذارد:
- جهشهای کدون توقف زودرس (PTC): با کاهش طول عمر، خطر بالای رویدادهای آئورتی، اسکولیوز شدید، اما خطر کمتر جراحی اکتوپیا لنتیس مرتبط هستند.
- جهشهای in-frame با از دست دادن سیستئین: با خطر بالای دیسکسیون آئورت (73%) و اسکولیوز شدید (45%) همراه هستند.
- جهشهای in-frame با افزودن سیستئین: خطر کمتر دیسکسیون آئورت (29%) اما خطر بالای جراحی اکتوپیا لنتیس (48%) دارند.
- جهشهای اگزونهای 24-32: با فنوتیپهای شدید، بهویژه سندرم مارفان نوزادی، مرتبط هستند.
مشاوره ژنتیک
سندرم مارفان یک بیماری اتوزومال غالب است، به این معنی که هر فرزند یک فرد مبتلا 50% شانس به ارث بردن بیماری را دارد. مشاوره ژنتیک شامل:
- ارزیابی خطر: توضیح احتمال انتقال بیماری به فرزندان.
- آزمایش ژنتیکی: برای تأیید تشخیص در افراد مشکوک یا اعضای خانواده.
- گزینههای تشخیصی پیش از تولد: مانند آزمایش ژنتیکی پیش از کاشت (PGT) یا آمنیوسنتز.
- برنامهریزی خانوادگی: بحث در مورد گزینههای تولیدمثلی برای کاهش خطر انتقال.
مشاوره ژنتیک بهویژه برای خانوادههایی که سابقه بیماری دارند یا در مواردی که جهش de novo شناسایی شده است، حیاتی است.
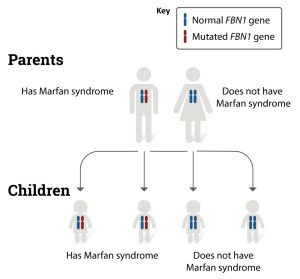
روشهای تشخیص ژنتیکی
روشهای اصلی تشخیص ژنتیکی عبارتند از:
- تحلیل توالی (Sequence Analysis): این روش برای شناسایی جهشهای نقطهای (missense، nonsense)، جهشهای splice site و حذف/درجهای کوچک در ژن FBN1 استفاده میشود. بر اساس منابع معتبر مانند GeneReviews، تحلیل توالی میتواند جهشهای بیماریزا را در حدود 93-90% افراد مبتلا به سندرم مارفان کلاسیک شناسایی کند. فناوریهای مورد استفاده شامل توالییابی نسل بعدی (Next-Generation Sequencing, NGS) است که امکان بررسی سریع و جامع ژن را فراهم میکند.
- تحلیل حذف/تکرار (Deletion/Duplication Analysis): حدود 5% موارد سندرم مارفان به دلیل حذفها یا تکرارهای بزرگتر در ژن FBN1 ایجاد میشوند که با تحلیل توالی قابل شناسایی نیستند. روشهایی مانند PCR کمی (quantitative PCR)، MLPA (Multiplex Ligation-dependent Probe Amplification) یا microarray هدفمند برای تشخیص این تغییرات استفاده میشوند.
- پانل چندژنی: در مواردی که تشخیص افتراقی با سایر اختلالات بافت همبند مانند سندرم لویز-دیتز یا سندرم اهلرز-دانلوس مورد نیاز است، پانلهای چندژنی که شامل FBN1 و ژنهای مرتبط مانند TGFBR1 و TGFBR2 هستند، استفاده میشوند. این پانلها معمولاً از NGS برای توالییابی و تحلیل حذف/تکرار استفاده میکنند.
- توالییابی کل اگزوم (Whole-Exome Sequencing, WES): در مواردی که فنوتیپ بیمار نامشخص است یا جهش در ژن FBN1 شناسایی نشده است، WES میتواند برای بررسی ژنهای دیگر مرتبط با بیماریهای مشابه استفاده شود. این روش، اگرچه جامعتر است، به دلیل هزینه بالا و پیچیدگی تفسیر نتایج، معمولاً به عنوان خط اول استفاده نمیشود.
استراتژیهای تشخیص ژنتیکی
بر اساس دستورالعملهایGeneReviews ، استراتژی استاندارد برای تشخیص ژنتیکی به شرح زیر است:
- آزمایش تکژنی FBN1: ابتدا تحلیل توالی ژن FBN1 انجام میشود. در صورت عدم شناسایی جهش، تحلیل حذف/تکرار انجام میشود. این رویکرد برای افرادی که معیارهای بالینی Ghent را دارند یا مشکوک به سندرم مارفان هستند، توصیه میشود.
- پانل چندژنی: در مواردی که تشخیص بالینی نامشخص است یا احتمال بیماریهای مشابه وجود دارد، پانلهای چندژنی که شامل FBN1 و ژنهای مرتبط هستند، ترجیح داده میشوند. این پانلها میتوانند خطر شناسایی جهشهای با اهمیت نامشخص (VUS) را کاهش دهند.
- توالییابی کل اگزوم یا ژنوم: این روشها در موارد نادر که هیچ جهشی در FBN1 یا ژنهای مرتبط یافت نشده است، استفاده میشوند. این رویکرد به دلیل پیچیدگی و هزینه بالا، کمتر رایج است.
نقش آزمایش ژنتیکی در تشخیص
آزمایش ژنتیکی در موارد زیر نقش کلیدی دارد:
- تأیید تشخیص: در افرادی که معیارهای بالینی Ghent را به طور کامل برآورده نمیکنند، شناسایی جهش بیماریزا در FBN1 میتواند تشخیص را تأیید کند.
- تشخیص افتراقی: کمک به تمایز سندرم مارفان از سایر اختلالات بافت همبند مانند سندرم لویز-دیتز یا سندرم اهلرز-دانلوس.
- مشاوره ژنتیک: شناسایی جهش امکان ارزیابی خطر برای اعضای خانواده و برنامهریزی تولیدمثلی (مانند تشخیص پیش از کاشت) را فراهم میکند.
- مدیریت بالینی: تشخیص زودهنگام میتواند به پایش منظم و مداخلات پیشگیرانه مانند درمان دارویی یا جراحی کمک کند.
دستورالعملهای بینالمللی برای تشخیص ژنتیکی
معیارهای Ghent نقش آزمایش ژنتیکی را در تشخیص سندرم مارفان برجسته میکنند. در غیاب سابقه خانوادگی، وجود جهش بیماریزا در FBN1 همراه با یک معیار اصلی (مانند اتساع ریشه آئورت یا اکتوپیا لنتیس) برای تشخیص کافی است. در حضور سابقه خانوادگی، شناسایی جهش FBN1 میتواند تشخیص را در افرادی با تظاهرات خفیف تأیید کند.
جدول 2: مقایسه روشهای تشخیص ژنتیکی سندرم مارفان
| روش | کاربرد اصلی | حساسیت تشخیصی | روش |
| تحلیل توالی FBN1 | تشخیص اولیه در افراد با معیارهای بالینی یا مشکوک | 93 – 90 درصد | تحلیل توالی FBN1 |
| تحلیل حذف/تکرار FBN1 | در صورت منفی بودن تحلیل توالی | 5 درصد | تحلیل حذف/تکرار FBN1 |
| پانل چندژنی | تشخیص افتراقی با سایر اختلالات بافت همبند | متغیر (بسته به آزمایشگاه) | پانل چندژنی |
| توالییابی کل اگزوم (WES) | در موارد پیچیده یا عدم شناسایی جهش در FBN1 | متغیر | توالییابی کل اگزوم (WES) |
| ARMS-PCR | آزمایش در اعضای خانواده | بالا برای جهشهای خاص | شناسایی جهشهای خاص در جمعیتهای بزرگتر |
درمان و مدیریت
مدیریت سندرم مارفان چند رشتهای است و بر پیشگیری از عوارض، بهویژه عوارض قلبی-عروقی، متمرکز است.
درمان دارویی
- بتابلوکرها: مانند آتنولول (4-1 میلیگرم/کیلوگرم/روز) برای کاهش فشار بر ریشه آئورت.
- مسدودکنندههای گیرنده آنژیوتانسین (ARBs) II: مانند لوزارتان (1.4-1.0 میلیگرم/کیلوگرم/روز) که میتواند اتساع آئورت را کاهش دهد. مطالعات نشان دادهاند که لوزارتان و آتنولول اثربخشی مشابهی دارند.
- ایربسارتان: تحقیقات اخیر نشان میدهد که ممکن است در کاهش اتساع آئورت مؤثرتر باشد.
مداخلات جراحی
- تعویض ریشه آئورت: توصیه میشود زمانی که قطر آئورت به ≥5.0 سانتیمتر برسد یا نرخ افزایش >0.5 سانتیمتر در سال باشد. روشهای valve-sparing مانند عمل دیوید اصلاحشده ترجیح داده میشوند.
- عمل PEARS: یک روش جدیدتر با خطر مرگومیر 1% در صورت انجام توسط جراحان با تجربه.
- جراحی چشمی: لنسکتومی برای اکتوپیا لنتیس شدید یا جراحی گلوکوم.
پایش
- اکوکاردیوگرافی: در زمان تشخیص، سپس هر 6 ماه تا پایداری، و پس از آن سالانه (یا هر 2-1 سال اگر z-score <+2.5).
- معاینات چشمی سالانه: از سن 1 سالگی برای بررسی اکتوپیا لنتیس و گلوکوم.
- پایش اسکلتی: معاینات هر 6 ماه برای اسکولیوز در سنین 18-6 سال.
مدیریت در کودکان
دستورالعملهای آکادمی اطفال آمریکا (AAP, 2023) توصیه میکنند:
- اکوکاردیوگرافی سالانه یا مکررتر در صورت z-score >+5.
- محدودیت فعالیتهای شدید مانند ورزشهای تماسی یا وزنهبرداری.
- معاینات چشمی سالانه از سن 1 سالگی.
- پایش اسکلتی برای اسکولیوز و ناهنجاریهای قفسه سینه.
تغییرات سبک زندگی
- اجتناب از ورزشهای پرشدت یا فعالیتهایی که فشار زیادی به آئورت وارد میکنند (مانند وزنهبرداری، غواصی).
- فعالیتهای کمفشار مانند پیادهروی، دوچرخهسواری یا شنا توصیه میشود.
- اجتناب از آنتیبیوتیکهای فلوروکینولون به دلیل خطر افزایش عوارض آئورتی.
آخرین تحقیقات و دستورالعملها
بر اساس منابع اخیر:
- BMJ Best Practice (2025): تأکید بر استفاده از بتابلوکرها و ARBs، و روشهای جراحی مانند عمل دیوید و PEARS.
- آکادمی اطفال آمریکا (2023): دستورالعملهای خاص برای کودکان، شامل پایش مکرر و محدودیتهای فعالیتی.
- تحقیقات در حال انجام: همکاریهایی مانند مطالعه دکتر سانجای سینها در دانشگاه کمبریج برای آزمایش داروهای جدید با استفاده از نمونههای پوستی بیماران برای ارزیابی تولید فیبریلین.
- مطالعات ایرانی: یک مطالعه در بیرجند (2023) یک جهش جدید در FBN1 را شناسایی کرد و از توالییابی کل اگزوم برای تشخیص استفاده کرد. این نشاندهنده دسترسی به فناوریهای پیشرفته ژنتیکی در ایران است.
در ایران، مدیریت سندرم مارفان بر اساس دستورالعملهای بینالمللی انجام میشود و آزمایش ژنتیکی در آزمایشگاههای تخصصی مانند آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی بوعلی قم در دسترس است.
آیا زندگی با سندرم مارفان چالشی سخت است یا میتوان به آن امید داشت؟
تشخیص زودهنگام سندرم مارفان میتواند تفاوت بزرگی در کیفیت زندگی فرد ایجاد کند. این بیماری اگر بهموقع شناسایی شود، بسیاری از علائم آن قبل از ایجاد مشکلات جدی، تحت کنترل قرار میگیرند. افراد مبتلا که تحت مراقبت منظم قرار میگیرند، میتوانند فعالیتهای روزمرهی خود را بدون نگرانی انجام دهند، شغل مورد علاقهی خود را دنبال کنند و زندگی عادی و طولانی داشته باشند. بنابراین، آگاهی از این بیماری و اقدام بهموقع برای کنترل آن، کلید داشتن یک زندگی سالم و بدون محدودیت است. علاوه بر مراقبتهای پزشکی، حمایت عاطفی خانواده نیز تأثیر عمیقی بر سلامت روحی و کیفیت زندگی فرد مبتلا دارد. فردی که با سندرم مارفان زندگی میکند، ممکن است به دلیل ظاهر متفاوت، محدودیتهای ورزشی، یا چالشهای پزشکی، احساس تنهایی یا ناامیدی داشته باشد. اما اگر خانواده و اطرافیان با درک، پذیرش و حمایت خود، فضای مناسبی را برای فرد فراهم کنند، اعتمادبهنفس او تقویت شده و مسیر زندگیاش هموارتر خواهد شد. همراهی خانواده در مراجعههای پزشکی، توجه به وضعیت سلامت فرد و ایجاد انگیزه برای پیشرفت، میتواند از استرس و نگرانیهای او بکاهد و انگیزهای برای زندگی بهتر ایجاد کند. بسیاری از افراد مبتلا به سندرم مارفان، با وجود این بیماری توانستهاند به موفقیتهای بزرگی در حوزههای مختلف دست یابند. فلورسک کوئین، والیبالیست مشهور آمریکایی، با وجود این بیماری در سطح حرفهای رقابت کرد. مورد کوئینتون کاکس، بازیکن فوتبال آمریکایی، علیرغم محدودیتهای فیزیکی به موفقیت ورزشی رسید. جان تاورنر، آهنگساز برجسته، بردفورد کاکس، خواننده و بازیگر، و جاویر بوتت، بازیگر مشهور، نیز با وجود ابتلا به مارفان، در دنیای هنر درخشیدند. این افراد ثابت کردهاند که سندرم مارفان مانعی برای موفقیت نیست، بلکه با آگاهی، مراقبت و اراده، میتوان نهتنها بر چالشهای آن غلبه کرد، بلکه زندگیای پربار و الهامبخش داشت.

نتیجهگیری
سندرم مارفان یک بیماری ژنتیکی پیچیده است که نیاز به تشخیص زودهنگام و مدیریت چند رشتهای دارد. با پایش منظم، درمان دارویی و مداخلات جراحی بهموقع، افراد مبتلا میتوانند زندگی طولانی و سالمی داشته باشند. مشاوره ژنتیک و آزمایش ژنتیکی نقش مهمی در مدیریت خانوادههای در معرض خطر دارند.
Reference:
http://BMJ Best Practice: Marfan syndrome
http://StatPearls: Marfan Syndrome
http://AAP: Health Supervision for Children and Adolescents with Marfan Syndrome
http://Clinical and genetic screening in a large Iranian family with Marfan syndrome
http://GeneReviews: FBN1-Related Marfan Syndrome
http://Tehran’s First Genetic Center Launched
http://Genetics and genomic medicine in Iran
https://doctorghavidel.ir/marfan-syndrome/
https://www.geniranlab.ir/%D8%B3%D9%86%D8%AF%D8%B1%D9%88%D9%85-%D9%85%D8%A7%D8%B1%D9%81%D8%A7%D9%86/
https://doctoreto.com/blog/what-is-marfan-syndrome/
https://diakoclinic.com/marfan-syndrome/