
مقدمه
آتروفی عضلانی نخاعی (SMA)[1] یک اختلال ژنتیکی است که به طور قابلتوجهی بر قدرت عضلانی و عملکردهای حرکتی افراد تأثیر میگذارد. این مقاله با هدف بررسی جامع بیماری SMA، مبانی ژنتیکی، علائم بالینی، روش های تشخیصی و گزینههای درمانی موجود تدوین شده است تا درک عمیقتری از این بیماری را برای خانوادهها و مراقبان افراد مبتلا فراهم کند.
بیماری SMA چیست؟
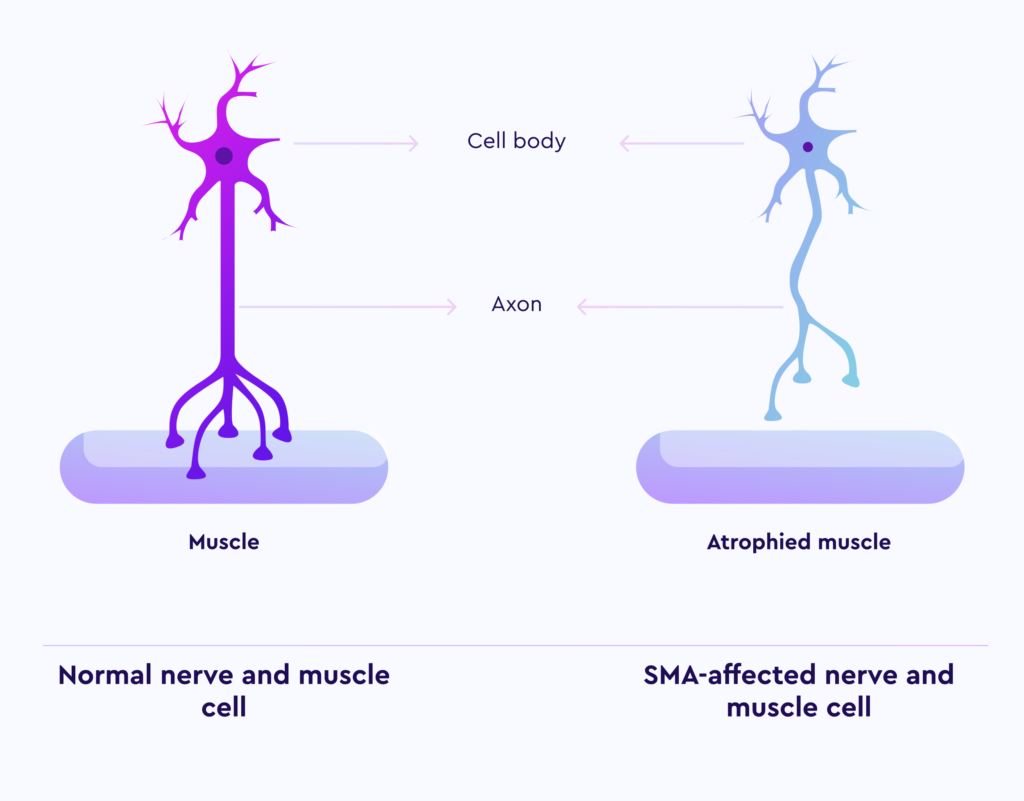
بیماری SMA اختلالی عصبی-عضلانی با منشأ ژنتیکی است که به طور تدریجی باعث از دست رفتن نورونهای حرکتی در فرد میشود. این نورونها وظیفه ارسال پیام عصبی به عضلات را بر عهده دارند و حرکتهای ارادی بدن وابسته به عملکرد آنها است. با آسیب دیدگی یا نابودی این نورونها قدرت حرکات ارادی کاهش یافته و در موارد شدید به طور کلی از بین میرود. تأثیر بیماری SMA بر نورونهای حرکتی سبب اختلال عملکرد در فعالیتهای حیاتی نظیر راه رفتن، تغذیه و تنفس میشود و در صورت عدم درمان، انواع شدید این بیماری میتوانند زندگی بیمار را تهدید کنند. در این بیماری، نورونهای حرکتی موجود در شاخ قدامی نخاع و بخش پایینی مغز دچار تحلیل و تخریب میشوند، که این امر موجب اختلال در انجام حرکات ارادی بدن مبتلایان میگردد. از لحاظ اپیدمیولوژیک، بیماری SMA پس از فیبروز کیستیک، دومین اختلال اتوزومال مغلوب شایع محسوب میشود و در ردهبندی بیماریهای مرتبط با تخریب عضلات نیز پس از دیستروفی عضلانی دوشن در جایگاه دوم قرار دارد. بررسیهای آماری نشان داده که احتمال ناقل بودن ژن معیوب مرتبط با بیماری SMA در جمعیتهای مختلف، در حدود یک نفر از هر 25 تا 50 نفر متغیر است. شیوع این بیماری در میان جمعیت عمومی نیز تقریباً یک مورد در هر 50,000 تا 100,000 تولد تخمین زده میشود. علاوه بر این، بر اساس تحقیقات، حدود یک نوزاد از هر شش هزار تولد غیر فامیلی و یک نوزاد از هر هزار تولد حاصل از ازدواجهای خویشاوندی نزدیک (مانند موارد ازدواج میان پسرعمو و دخترعمو یا پسرخاله و دخترخاله) به این بیماری مبتلا میشوند. همچنین مطالعات نشان میدهد که از هر 40 نفر یک نفر ناقل ژن معیوب مرتبط با این بیماری است و در صورتی که دو فرد ناقل ازدواج کنند، احتمال تولد فرزند مبتلا وجود خواهد داشت. با توجه به شیوع بالای ازدواجهای خویشاوندی در ایران و عدم وجود برنامههای غربالگری مناسب، احتمالاً میزان ابتلا در کشور بیش از میانگین جهانی است. بر اساس برآوردهای اولیه، تعداد بیماران مبتلا به SMA در ایران حداقل بین 3 تا 6 هزار نفر تخمین زده میشود.
انواع بیماری SMA
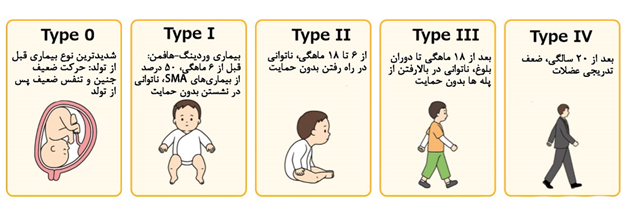
بیماری SMA به عنوان یکی از بیماریهای نوروژنتیکی مهم، به پنج نوع اصلی طبقهبندی میشود که هر یک ویژگیها و شدت متفاوتی دارند:
- نوع 1 (شدید): این نوع، شدیدترین فرم بیماری است و علائم آن معمولاً پیش از ششماهگی بروز میکنند. نوزادان مبتلا اغلب با مشکلات جدی در تنفس و بلع مواجه هستند و نیاز به مراقبتهای ویژه دارند.
- نوع 2 (متوسط): این نوع معمولاً بین 7 تا 18 ماهگی آغاز میشود. کودکان مبتلا قادر به راهرفتن نیستند و محدودیت در حرکتهای پایهای دارند.
- نوع 3 (خفیف): علائم این نوع پس از 18 ماهگی ظاهر میشوند و در مقایسه با انواع دیگر، شدت کمتری دارند. بیماران در این دسته معمولاً توان حرکتی بیشتری دارند اما ممکن است با ضعف تدریجی در فعالیتهای عضلانی مواجه شوند.
- نوع 4 (آغاز در بزرگسالی): این نوع بسیار نادر است و علائم آن در دوران بزرگسالی نمایان میشود. شدت علائم در این نوع معمولاً کم است.
- SMA ناشی از جهشهای نادر ژنتیکی: برخی موارد SMA به دلیل جهشهایی فراتر از الگوی معمول ژنتیکی ایجاد میشوند که ماهیتشان پیچیدهتر و کمتر شناخته شده است.
علائم بالینی بیماری SMA
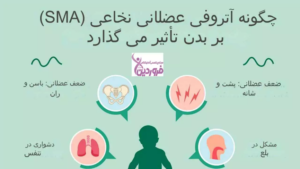
شناخت زودهنگام علائم بیماری SMA برای آغاز سریع مداخلات درمانی بسیار حائز اهمیت است. علائم عمومی بیماری شامل موارد زیر هستند:
- ضعف پیشرونده عضلانی
- ضعف و ناتوانی در کنترل حرکات
- مشکلات تنفسی و بلعی که با افزایش سن تشدید میشوند
- خستگی مفرط در انجام فعالیتهای روزمره
در دوره نوزادی، مهمترین نشانهها شلشدگی اندامها و تأخیر آشکار در مراحل طبیعی رشد، نظیر دیر نشستن یا غلتزدن، هستند. این شاخصها نیازمند بررسی دقیق توسط متخصصین جهت تشخیص زودهنگام بیماری و برنامهریزی برای درمان مناسب خواهند بود.
اساس ژنتیکی بیماری SMA
ژنها حامل اطلاعات ژنتیکی مورد نیاز برای توسعه و عملکرد بدن هر انسان هستند. بیماری SMA ناشی از جهش در دو ژن اصلی به نامهای SMN1 و SMN2 میباشد:
- ژن SMN1: به عنوان ژن اصلی تولیدکننده پروتئین بقا دهنده نورون حرکتی (SMN)[2] شناخته شده و بر روی کروموزوم شماره 5 قرار دارد.
- ژن SMN2: این ژن به عنوان “ژن مکمل” فعالیت میکند و پروتئین SMN را با بهرهوری پایینتری نسبت به ژن SMN1 تولید میکند.
بیماری SMA معمولاً با الگوی وراثت اتوزومی مغلوب منتقل میشود. اغلب افراد دو نسخه از ژن SMN1 دارند که یکی از هر یک از والدین به ارث میرسد. در کودک مبتلا به SMA، دو نسخه ناقص یا معیوب از ژن SMN1 از والدینی که معمولاً ناقل سالم این ژن هستند، به ارث میرسند. از این رو، وجود یک نسخه معیوب (هتروزیگوت) معمولاً منجر به بروز علائمی نمیشود.
جهش در ژن SMN1 عمدتاً منجر به بروز چهار نوع اصلی بیماری SMA (نوع 1، 2، 3 و 4) میشود. این جهشها به کوتاهتر شدن پروتئین SMN و کاهش کارایی آن منجر میشوند. برخی از بیماران مبتلا به SMA در انواع 2، 3 و 4، دارای سه یا بیش از سه نسخه (رونوشت) از ژن SMN2 در هر سلول هستند که این نسخههای اضافی با تعدیل فنوتیپ بیماری، یک شکل خفیفتر از آن را ایجاد میکنند.
در بین بیماران SMA با شروع علائم در سنین خردسالی، حذف هموزیگوت اگزونهای 7 و 8 در ژن SMN1 در 95 تا 98 درصد موارد شایع است. در حالی که در یک تا دو درصد بیماران، حذف اگزونهای مذکور در یکی از آللها مشاهده نمیشود و جهش نقطهای مسئول علائم است.
یک فرم نادر بیماری SMA با الگوی وراثت اتوزومی غالب یا وابسته به کروموزوم X نیز وجود دارد. مثالی کلینیکی از این فرمها، بیماری کندی[3]یا آتروفی عضلانی نخاعی و بولبار[4] (BSMA, SBMA) است. این نوع از SMA برخلاف انواع رایج، ناشی از موتاسیون در ژن SMN1 نیست و کمبود پروتئین SMN نیز در آن مشاهده نمیشود. تفاوت دیگری که در این فرم قابل توجه است، درگیری عضلات دورتر از مرکز بدن است؛ بر خلاف انواع معمول SMA (1 تا 4) که غالباً عضلات نزدیکتر به محور مرکزی بدن را تحت تأثیر قرار میدهند.
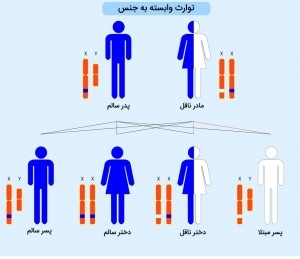
مشاوره ژنتیک بیماری SMA
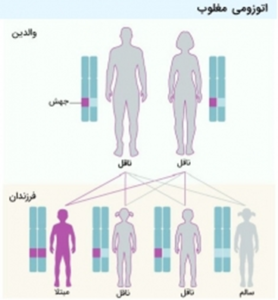
این بیماری به صورت اتوزومال مغلوب به ارث میرسد، به این معنا که در افراد مبتلا، هر دو نسخه ژن در هر سلول دچار جهش شدهاند. افرادی که تنها یک نسخه از ژن معیوب را دارند، حامل بیماری محسوب میشوند، اما عموماً علائم یا نشانههای بیماری را نشان نمیدهند و فنوتیپ سالمی دارند. در صورتی که زوجی یک فرزند مبتلا به SMA داشته باشند، احتمال وقوع نتایج زیر در بارداریهای آینده وجود دارد:
- حدود 25 درصد احتمال تولد یک کودک مبتلا.
- تقریبا 50 درصد احتمال تولد یک کودک حامل ژن معیوب بدون علائم.
- نزدیک به 25 درصد احتمال تولد یک کودک سالم.
در شرایط نادر، تخمین زده میشود که حدود 2 درصد از افراد مبتلا (ما می دانیم این افراد دو نسخه جهش یافته از ژن SMN1 دارند) ممکن است یک نسخه از ژن جهشیافته را به صورت جهشی تازه یا جدید[5] ایجاد کرده باشند. در چنین مواردی، تنها یکی از والدین حامل واریانت ژنتیکی مرتبط با ژن SMN1 خواهد بود و در نتیجه، چون والد دیگر سالم است، احتمال به ارث رسیدن این بیماری به سایر فرزندان خانواده افزایش نمییابد.
تشخیص بیماریSMA
تشخیص اولیه بیماری SMA از طریق معاینات بالینی توسط پزشک متخصص صورت میپذیرد که شامل انجام تستهای فیزیکی، بررسی نوارهای عصبی و الکترومیوگرام و همچنین مطالعه سوابق خانوادگی بیمار است. برای تأیید نهایی این بیماری، آزمایشهای ژنتیکی با استفاده از روشهای مولکولی مورد نیاز است.
تشخیص ژنتیکی بیماری SMA
به دلیل ارثی بودن، این بیماری از اهمیت بالایی برخوردار است. در این راستا، ضروری است که تمامی اعضای خانوادههای درجه یک و دو، بهویژه پیش از تصمیمگیری برای ازدواج یا فرزندآوری، تحت آزمایشهای و بررسیهای ژنتیکی قرار گیرند. این گونه اقدامات نه تنها به شناسایی جهشها و نقصهای ژنتیکی موجود در خانواده کمک میکند، بلکه امکان پیشگیری از انتقال این بیماری به نسلهای آینده را نیز فراهم میآورد. بررسی خطر ابتلا به بیماری SMA در افراد، در دو مرحله قابل انجام است:
- بررسی ژنتیکی از طریق آزمایش خون: در این مرحله، از زوجین، والدین فرد مبتلا یا خود بیمار نمونه خون گرفته میشود تا وضعیت ناقل بودن یا ابتلا به بیماری ژنتیکی مشخص شود. این آزمایشها کمک میکنند تا در صورت وجود احتمال انتقال، اقدامات پیشگیرانه لازم صورت پذیرد.
- بررسی ژنتیکی در دوران بارداری: دو روش اصلی در دوران بارداری وجود دارد:
- آمنیوسنتز(AF)[6]: این روش معمولاً بین هفتههای 14 تا 18 بارداری انجام میشود. در این آزمایش، نمونهای از مایع آمنیوتیک با کمک یک سوزن از رحم گرفته میشود. مایع به دست آمده حاوی سلولهای جنینی است که از نظر وجود جهشهای مرتبط با SMA مورد بررسی قرار میگیرند.
- نمونهبرداری از پرزهای کوریونی (CVS)[7]: این آزمایش معمولاً بین هفتههای 10 تا 13 بارداری انجام میشود. در این روش، نمونهای از پرزهای کوریونی که بهصورت برآمدگیهای انگشتمانند روی جفت قرار دارند، از طریق دهانه رحم یا دیواره شکم جمعآوری میشود. این نمونه نیز حاوی سلولهای جنینی است و برای شناسایی جهشهای ایجادکننده SMA بررسی میشود.
اجرای این فرآیندها و اهمیت به اقدامات پیشگیرانه میتواند نقش مؤثری در کاهش شیوع بیماری و بهبود کیفیت زندگی نسلهای آینده داشته باشد.
شایان ذکر است که تشخیص ژنتیکی این بیماری از طریق روشهای مولکولی و با استفاده از کیت MLPA[8] اختصاصی برای بیماری SMA در آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی بوعلی قم امکانپذیر است .
ﻏﺮﺑﺎﻟﮕﺮی ژنتیکی بیماری SMA
به طور کلی، آزمایشهای ژنتیکی غربالگری برای بررسی احتمال وجود ژنهای معیوب مرتبط با بیماریهای شناختهشده ژنتیکی طراحی شدهاند. با انجام این آزمایشها، چه پیش از بارداری و چه در طول آن، میتوان میزان خطر ابتلای فرزند به بیماریهای ژنتیکی را ارزیابی کرد. انجام آزمایشهای پیش از تولد برای بارداریهایی که بهواسطه عوامل گوناگون پرخطر محسوب میشوند، ضروری بوده و نقشی کلیدی در پیشبینی و پیشگیری از انتقال بیماری به نسل بعد دارد. یکی از این آزمایشها، تست غربالگری بیماری SMA است که احتمال ابتلای فرزند به این بیماری را پیشبینی میکند. این آزمایش روی نمونه خون انجام میشود و معمولاً ابتدا یکی از والدین مورد بررسی قرار میگیرد. در صورتی که نتایج نشان دهد والد مذکور ناقل است، آزمایش روی والد دیگر نیز انجام میشود. گزارش نتایج این تست براساس تعداد نسخههای طبیعی ژن SMN1 ارائه میشود. اگر فردی دارای دو یا بیشتر از دو نسخه سالم از این ژن باشد، احتمال ناقل بودن او بسیار پایین است. در مقابل، اگر تنها یک نسخه سالم از ژن SMN1 وجود داشته باشد، نسخه دیگر معیوب در نظر گرفته شده و فرد ناقل بیماری SMA محسوب میشود.
با توجه به شیوع بالای ناقلین بیماری SMA در ایران، انجام این آزمایش به تمامی زوجهایی که در مراحل اولیه تصمیمگیری برای بارداری هستند یا در سهماهه اول بارداری قرار دارند، توصیه میشود. بهترین زمان برای انجام این آزمایش قبل از بارداری است؛ چرا که در صورت وجود نتایج مثبت، فرصت لازم برای اقدامات لازم و برنامهریزیهای بعدی فراهم خواهد بود.
تشخیص پیش از تولد و تشخیص ژنتیکی قبل از لانهگزینی
برای شناسایی بیماری SMA، دو روش قابل پیشنهاد به والدین است:
- تشخیص ژنتیکی پیش از تولد
- تشخیص پیش از لانهگزینی
در صورتی که یک عامل بیماریزا در هر دو والد شناسایی شده باشد، احتمال ابتلای فرزند دیگر به بیماری ۲۵ درصد خواهد بود. اما در صورت وجود یک عامل بیماریزا، احتمال تکرار بیماری کمتر برآورد میشود.
گزینههای درمانی برای بیماری SMA
در حالی که درمان قطعی وجود ندارد، پیشرفتهای قابل توجهی در درمانهای SMA به وجود آمده است:
- Nusinersen (Spinraza): این یک درمان تایید شده توسط FDA است که اتصال SMN2 را برای افزایش تولید پروتئین SMN اصلاح می کند. بیماران مبتلا از طریق تزریق داخل نخاعی ممکن است بهبودهایی در عملکرد حرکتی نشان دهند.
- Onasemnogene abeparvovec-xioi (Zolgensma): یک رویکرد ژن درمانی که یک کپی عملکردی از ژن SMN1 را به سلول های بیمار تحویل می دهد و به طور بالقوه یک گزینه درمانی برای نوزادان مبتلا به بیماری SMA را ارائه می دهد.
- Risdiplam (Evrysdi): یک داروی خوراکی که سطح SMN را در بدن افزایش می دهد و برای بیماران در تمام سنین تایید شده است.
- درمان های حمایتی: در کنار این درمان ها، خدمات توانبخشی شامل فیزیوتراپی، کاردرمانی و مراقبت های تنفسی برای مدیریت علائم و بهبود کیفیت زندگی حیاتی هستند.

زندگی با بیماری SMA
زندگی با بیماری SMA چالشهای خاصی را به همراه دارد. یک سیستم حمایتی قوی برای بیماران و مراقبان ضروری است.
جمع بندی
بیماری SMA یک وضعیت پیچیده است، اما پیشرفتهای تحقیقاتی امیدوارکننده است. تشخیص زودهنگام و درمانهای پیشرفته میتواند به طور چشمگیری نتایج را بهبود بخشد. افزایش آگاهی در مورد بیماری SMA برای کمک به خانوادهها در شناخت علائم اولیه و دسترسی به منابع حیاتی است. آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی بوعلی قم برای کمک به خانواده ها، یک خدمت جامع شامل انواع آزمایش های ژنتیکی برای بیماری SMA ارائه می دهد.
[1] Spinal Muscular Atrophy
[2] Survival of Motor Neuron
[3] Kennedy Disease
[4] Spinal and bulbar (bulbospinal) muscular atrophy
[5] De novo
[6] Amniocentesis
[7] Chorionic Villus Sampling
[8] Multiplex Ligation-Dependent Probe Amplification
Reference: