مقدمه
سندرم نیمن پیک Niemann-Pick Disease) ، (NPD یک گروه نادر از اختلالات ژنتیکی است که تحت عنوان بیماریهای ذخیرهای لیزوزومی شناخته میشود. این اختلالات بهطور عمده با نقص در متابولیسم لیپیدها، بهویژه اسفنگومیلین و کلسترول، همراه هستند که منجر به تجمع این مواد در سلولهای گوناگون بدن از جمله کبد، طحال، مغز و ریهها میشود. این انباشتگی به مرور زمان به آسیبهای جدی ارگانها و سیستم عصبی منجر میشود. سندرم نیمن پیک بر اساس منشأ ژنتیکی و علائم بالینی به سه نوع اصلی A، B و C تقسیم شده است، در حالی که نوع D پیشتر زیرمجموعهای از نوع C در نظر گرفته میشد. شیوع کلی این بیماری بین ۱ در ۱۵۰,۰۰۰ تا ۲۵۰,۰۰۰ تولد تخمین زده میشود، اما در برخی جمعیتهای خاص مانند یهودیان اشکنازی، نوع A بهطور قابل توجهی شایعتر است. مطالعات اخیر بینالمللی نشاندهنده پیشرفتهای قابل توجه در زمینه تشخیص زودهنگام و مدیریت بیماری است. این پیشرفتها شامل استفاده از بیومارکرهای پلاسما مانند اکسیاسترولها و لیزواسفنگومیلینها بوده که دقت بالایی در فرایند غربالگری دارند. در ایران نیز بررسیهایی در زمینه تشخیص مولکولی و بالینی انجام شده که موارد پراکنده این بیماری با تمرکز بر جهشهای ژنتیکی خاص در جمعیت ایرانی را نمایان کرده است.
علائم سندرم نیمن پیک
علائم عمومی ممکن است بسته به نوع بیماری متفاوت باشند، اما رایجترین نشانهها شامل موارد زیر هستند:
- هپاتواسپلنومگالی (بزرگی کبد و طحال)
- مشکلات ریوی (مانند ابتلا به عفونتهای مکرر)
- اختلالات عصبی (مانند تأخیر در رشد، تشنج، یا مشکلات بلع)
- لکه قرمز گیلاسی در چشم (مشاهده در ۵۰ درصد موارد نوع A)
- بر اساس مطالعات اخیر، توجه ویژهای به علائم اولیه مانند کلستاز نوزادی (زردی) در نوع C شده است.
انواع سندرم نیمن پیک
سندرم نیمن پیک بر اساس ژنهای دخیل و علائم بالینی به انواع زیر دستهبندی میشود:
- نوع A: شدیدترین نوع بیماری که معمولاً در دوران نوزادی بروز میکند. این نوع ناشی از کمبود کامل آنزیم اسید اسفنگومیلیناز (ASM) بوده و با بزرگ شدن کبد و طحال (هپاتواسپلنومگالی)، مشکلات ریوی و آسیبهای عصبی همراه است. امید به زندگی در این افراد معمولاً کمتر از ۲ تا ۳ سال است.
- نوع B: نسبت به نوع A خفیفتر بوده و حاصل کمبود جزئی ASM است. علائم اصلی شامل بزرگی کبد و طحال و مشکلات ریوی بوده اما عوارض عصبی کمتر شایع هستند. افراد مبتلا معمولاً تا بزرگسالی زنده میمانند.
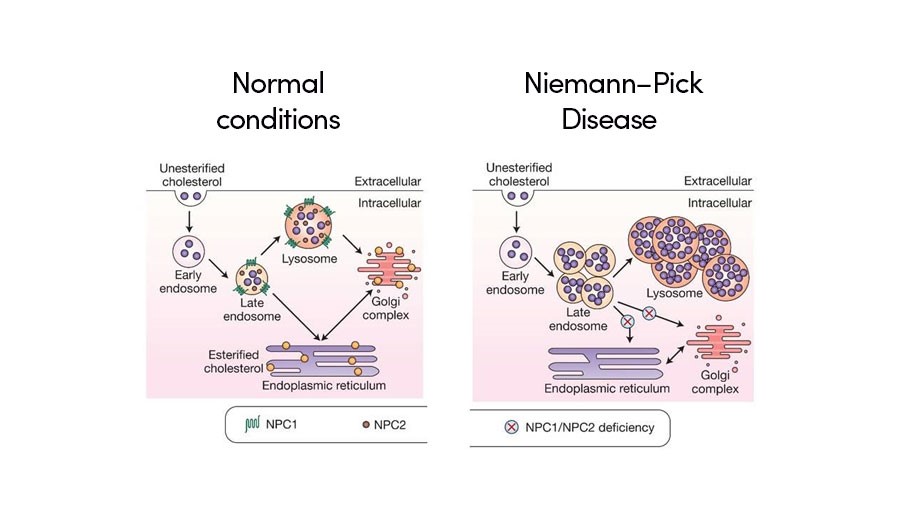
- نوع C: این نوع ناشی از اختلال در حمل کلسترول داخل سلولی است که توسط ژنهای NPC1 یا NPC2 کنترل میشود. علائم شامل مشکلات عصبی همچون آتاکسی، دیستونی و دشواری در بلع بوده و سن شروع بیماری میتواند از نوزادی تا بزرگسالی متغیر باشد. نوع C1 بیشترین موارد را شامل میشود (حدود ۹۵ درصد).
در ایران، گزارشهایی از موارد مربوط به انواع A و B منتشر شده است و مطالعات عموماً بر تشخیص مولکولی در خانوادههایی با سابقه ژنتیکی تمرکز دارند.
اساس ژنتیکی سندرم نیمن پیک
اساس ژنتیکی سندرم نیمن پیک نشاندهنده یک بیماری اتوزومال مغلوب است، به این معنا که برای بروز آن، فرد باید دو نسخه جهشیافته از ژن مرتبط را از والدین به ارث ببرد. والدینی که حامل (هتروزیگوت) هستند، عموماً بدون علائم بالینی هستند، اما احتمال ابتلای فرزند به این بیماری ۲۵ درصد است.
- انواع A و B:
این دو نوع در اثر جهشهایی در ژن SMPD1، واقع بر کروموزوم 11p15، ایجاد میشوند. این ژن مسئول کدگذاری آنزیم اسید اسفنگومیلیناز (ASM) است. تاکنون بیش از ۱۸۰ جهش مختلف در این ژن شناسایی شدهاند که شامل جهشهای بدمعنی (۶۰ درصد)، تغییر قالب (۱۹ درصد)، بی معنی و حذفها میباشد. در نوع A، جهشها موجب کاهش کامل فعالیت آنزیم ASM میشوند؛ نمونههایی از این جهشها عبارتاند از R۴۹۶L، L۳۰۲P که در جمعیت اشکنازی شایع هستند. در نوع B، فعالیت باقیمانده آنزیم بین ۵ تا ۲۰ درصد گزارش شده است؛ جهشهایی مانند حذفهای خفیف از این دسته هستند.
- نوع C:
این نوع ناشی از جهشهایی در ژنهای NPC۱ (روی کروموزوم ۱۸ که عامل ۹۵ درصد موارد است) یا NPC۲ (روی کروموزوم ۱۴) است. وظیفه این ژنها تنظیم حملونقل کلسترول درون سلولی است. تاکنون بیش از ۴۰۰ جهش در ژن NPC۱ شناسایی شده است.
تشخیص آزمایشگاهی (بیوشیمیایی)
تشخیص آزمایشگاهی و ژنتیکی در زمینه بیماریهایی مانند نیمن پیک نقش حیاتی دارد و با استفاده از روشهای بیوشیمیایی و ژنتیکی دقیق، امکان تشخیص زودهنگام و مؤثر فراهم میشود.
تشخیص اولیه معمولاً با توجه به علائم بالینی نظیر هپاتواسپلنومگالی شروع شده و سپس آزمایشهای بیوشیمیایی برای تأیید صورت میگیرد. پیشرفتهای اخیر شامل استفاده از بیومارکرهای حساس برای تشخیص زودهنگام بوده است:
- اندازهگیری فعالیت ASM: در انواع A و B، فعالیت آنزیم در لکوسیتهای خون بررسی میشود. نوع A کاهش کامل فعالیت (<1%) داشته، در حالی که نوع B دارای کاهش جزئی (5-20%) است.
- بیومارکرها: لیزواسفنگومیلین (Lyso-SM) و Lyso-SM-509 بهعنوان نشانگرهای تفکیکدهنده بین انواع A/B و C کاربرد دارند. برای نوع C، اکسیاسترولها مانند cholestane-3β,5α,6β-triol مورد استفاده قرار میگیرند.
- بیوپسی و رنگآمیزی: از رنگآمیزی فیلیپین جهت شناسایی تجمع کلسترول در فیبروبلاستها برای تشخیص نوع C استفاده میشود و سلولهای فوم نیز با آسپیراسیون مغز استخوان بررسی میشوند.
- تصویربرداری و آزمایشهای جانبی: MRI برای بررسی مغز، کبد و طحال و معاینات چشم تخصصی جهت شناسایی VSGP در نوع C انجام میشود.
در ایران، یکی از مشکلات اصلی تأخیر در تشخیص است. استفاده از روش dried blood spot برای اندازهگیری Lyso-SM-509 جهت تسریع فرایند تشخیص توصیه شده است.
تشخیص آزمایشگاهی (ژنتیکی)
تشخیص ژنتیکی ابزار کلیدی در تأیید بیماری و شناسایی حاملان بوده و تکنیکهای پیشرفتهای مانند NGS و WES برای آن به کار گرفته میشود.
- توالییابی ژنتیکی: توالییابی Sanger و NGS جهت شناسایی جهشهای مرتبط با ژن SMPD1 (انواع A/B) یا NPC1/NPC2 (نوع C) کاربرد دارند. در موارد پیچیده مانند جهش نوظهور 2925_2928delCTGC در NPC1، استفاده از WES ضروری است.
- تست پیشتولدی: سلولهای آمنیوسیت یا ویلوس کوریونی برای بررسی آنزیم یا تحلیل مولکولی استفاده میشوند.
- غربالگری cffDNA: این روش که بر اساس DNA جنینی موجود در خون مادر انجام میشود، بهعنوان روشی نوین برای تشخیص جهشهای NPC1 در دوران بارداری به شمار میرود.
مطالعات در ایران نشان دادهاند که ۱۰ جهش نوظهور در ۱۹ بیمار وجود داشته است. استفاده از روش توالییابی Sanger همراه با تحلیلهای پیشرفته بیوانفورماتیک برای پیشبینی شدت بیماری پیشنهاد شده است.
مشاوره ژنتیک
مشاوره ژنتیک در خانوادههای پرریسک، بهویژه در ایران با نرخ بالای ازدواجهای فامیلی (تا حدود ۶۰ درصد)، اهمیت بالایی دارد. این فرآیند شامل ارزیابی میزان خطر، ارائه گزینههای پیشگیرانه و حمایت روانی است.
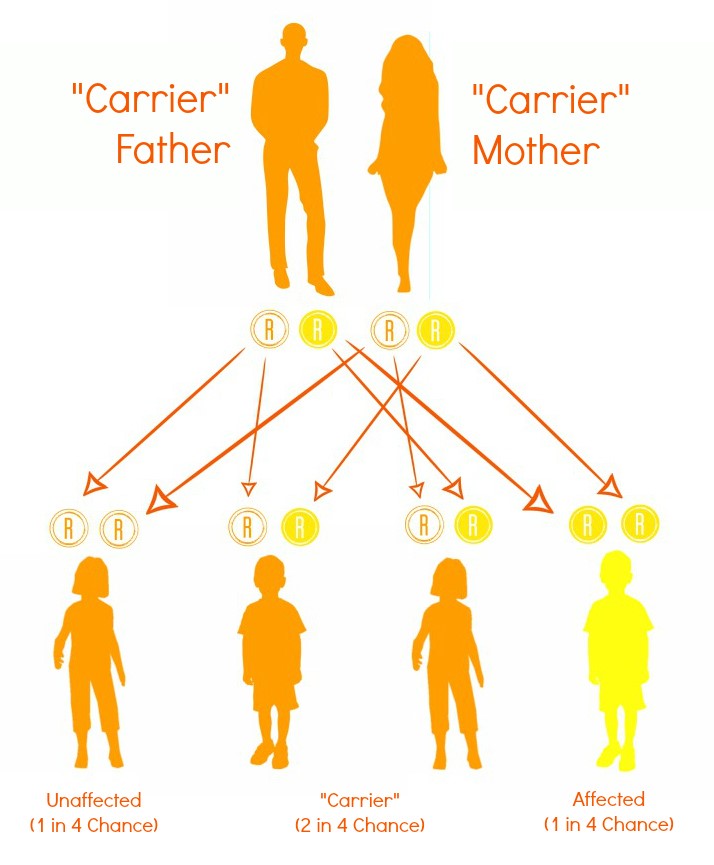
- آزمایش حاملگی: برای والدینی که احتمال وجود جهش ژنتیکی در ژنهای SMPD1 یا NPC1 دارند. در صورتی که هر دو والدین حامل باشند، خطر بروز بیماری در فرزند تا ۲۵ درصد خواهد بود.
- بررسیهای پیش از تولد: شامل آزمایش ژنتیکی در دوران لقاح مصنوعی (IVF) از طریق PGT یا تشخیصیهای جنینی نظیر نمونهبرداری از پرزهای جفتی (CVS) و آمنیوسنتز برای شناسایی ناهنجاریهای احتمالی.
- مشاوره خانوادگی: بحث درباره روشهای درمان حمایتی مانند میگلوسات در بیماری C و همچنین ارزیابی مخاطرات مرتبط با حاملگی. در ایران، مراکز تخصصی ژنتیکی عمدتاً بر غربالگریهای ژنتیکی پیش از ازدواج تمرکز دارند و از روشهای تشخیصی پیش از تولد نیز استفاده میشود.
این خدمات میتوانند نقش مؤثری در کاهش ریسک انتقال بیماریهای ژنتیکی به نسل بعد و بهبود سلامت خانوادهها داشته باشند.
| مرحله مشاوره | اقدامات پیشنهادی | ریسک مرتبط |
| پیش از ازدواج | تست حاملگی ژنتیکی | کاهش ریسک در ازدواج فامیلی |
| بارداری | غربالگری cffDNA یا CVS | تشخیص زودرس جنینی |
| پس از تشخیص | حمایت روانی و برنامهریزی خانوادگی | مدیریت ریسک برای فرزندان بعدی |
آخرین پیشرفتها و درمان
بر اساس مقالات اخیر، درمان اصلی از نوع حمایتی است، اما میگلوسات برای نوع C (کاهش علائم عصبی) تأیید شده است. همچنین ژندرمانی در مدلهای حیوانی امیدوارکننده ظاهر شده است. با این حال، در ایران، تمرکز بر تشخیص زودرس برای بهبود کیفیت زندگی است.
نتیجهگیری
سندرم نیمن پیک با پیشرفتهای ژنتیکی و تشخیصی، قابل مدیریتتر شده است. تشخیص زودرس از طریق بیومارکرها و WES، همراه با مشاوره ژنتیک، میتواند کیفیت زندگی را بهبود بخشد. برای مشاوره، با متخصصان ژنتیک تماس بگیرید.
Reference:
https://www.ncbi.nlm.nih.gov/books/NBK1370/
https://www.nature.com/articles/s41436-020-01043-7
https://www.jci.org/articles/view/160900
https://www.cell.com/ajhg/fulltext/S0002-9297(21)00234-5
https://www.nejm.org/doi/full/10.1056/NEJMra2000758
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)31725-0/fulltext
https://www.sciencedirect.com/science/article/pii/S1096719221003456
https://www.genetics.org/content/216/2/431
https://www.frontiersin.org/articles/10.3389/fgene.2021.678155/full