مقدمه
سندرم برنارد-سولیر (BSS) یک اختلال نادر خونی ارثی است که فرآیند لخته شدن خون را مختل میکند و خطر خونریزیهای شدید را افزایش میدهد. این بیماری اغلب از دوران نوزادی یا کودکی آشکار میشود و با علائمی مانند کبودیهای آسان، خونریزی بینی مکرر و دورههای قاعدگی سنگین همراه است. بر اساس گزارشهای اخیر، شیوع جهانی BSS حدود ۱ در ۱ میلیون نفر تخمین زده میشود، اما در ایران، به دلیل عوامل ژنتیکی و الگوهای ازدواج خویشاوندی، شیوع آن بالاتر است و مطالعات موارد متعددی را ثبت کردهاند.
تاریخچه و اپیدمیولوژی سندرم برنارد-سولیر
سندرم برنارد-سولیر برای اولین بار در سال ۱۹۴۸ توسط پزشکان فرانسوی، ژان برنارد و ژان-پییر سولیر، توصیف شد. آنها این اختلال را بر اساس مشاهدات بالینی از بیماران با ترومبوسیتوپنی (کاهش پلاکت) و پلاکتهای غولپیکر شناسایی کردند. از آن زمان، BSS به عنوان یکی از شایعترین اختلالات عملکرد پلاکتی ارثی شناخته شده است.
از نظر اپیدمیولوژی، BSS در سراسر جهان نادر است، اما در جوامع با نرخ بالای ازدواجهای خویشاوندی مانند جنوب ایران، شیوع آن قابل توجهتر است. یک مطالعه در تهران بر روی ۹۷ بیمار نشان داد که ۶۳.۹% آنها با خونریزی بینی (اپیستاکسی) مراجعه کردهاند و ۶۸.۷% پلاکتهای غولپیکر در لام خونی داشتهاند. در جنوب ایران، شیوع BSS و اختلالات مشابه مانند ترومبوآستنی گلانزمن بالاتر از میانگین جهانی گزارش شده است. در سال ۱۴۰۴، گزارشهای موردی از مراکش و چین نیز جهشهای جدیدی را برجسته کردهاند که بر اهمیت غربالگری ژنتیکی تأکید دارند.
سندرم برنارد-سولیر چیست؟
BSS یک اختلال اتوزومال مغلوب است که پلاکتهای خون را تحت تأثیر قرار میدهد. پلاکتها در افراد مبتلا تعداد کمتری (کمتر از ۱۵۰,۰۰۰ در میکرولیتر) دارند، اندازهشان بزرگتر است و عملکرد چسبندگیشان به دیواره رگها مختل میشود. این مشکل به دلیل کمبود کمپلکس گلیکوپروتئین Ib-IX-V رخ میدهد که نقش کلیدی در اتصال پلاکتها به عامل فون ویلبراند (vWF) دارد.
علاوه بر نوع ارثی، موارد اکتسابی BSS نیز گزارش شده که اغلب با عفونتها، کمبود GATA2 یا مهارکنندههای پلاسمایی مرتبط است. برای مثال، مواردی از BSS اکتسابی به عنوان عارضه سل ریوی یا اختلالات ایمنی توصیف شده است.
علائم سندرم برنارد-سولیر
علائم BSS معمولاً از بدو تولد یا کودکی ظاهر میشود و شدت آن متغیر است. شایعترین علائم عبارتند از:
- خونریزی بینی مکرر و طولانی: شایعترین علامت در ۶۰-۷۰% موارد که ممکن است بدون ضربه رخ دهد.
- کبودی آسان و پتشی: لکههای قرمز یا بنفش زیر پوست پس از ضربههای جزئی.
- خونریزی از لثه یا جراحات کوچک: بریدگیهای ساده میتواند ساعتها طول بکشد.
- منوراژی (خونریزی قاعدگی سنگین): در زنان که میتواند به کمخونی منجر شود.
- علائم شدیدتر: خونریزی گوارشی، هماتوری (خون در ادرار) یا خونریزی پس از جراحی که در ۴۰% بیماران حداقل یک بار رخ داده است.
در کودکان، BSS اغلب با علائم خفیف شروع میشود، اما در بزرگسالی میتواند تشدید یابد، به ویژه در بارداری یا عفونتها.
علل و مکانیسمهای مولکولی
علت اصلی BSS جهش در ژنهای GP1BA، GP1BB یا GP9 است که پروتئینهای کمپلکس GPIb-IX-V را کد میکنند. این جهشها ارثی و اتوزومال مغلوب هستند، به این معنا که هر دو والد باید حامل باشند.
تحقیقات اخیر جهشهای جدیدی مانند هتروزیگوت ترکیبی در GP1BA را شناسایی کردهاند و نقش مسیر RhoA در فعالسازی پلاکتها را برجسته کردهاند. این مسیر که توسط αIIbβ3/SRC تنظیم میشود، در شدت خونریزی مؤثر است. در ایران، مطالعات ژنتیکی بر روی بیماران BSS جهشهای محلی را تأیید کرده و بر ارتباط با ازدواجهای خویشاوندی تأکید دارند.
روشهای تشخیص
تشخیص BSS بر اساس ترکیبی از تاریخچه بالینی، آزمایشهای خونی و ژنتیکی است:
- آزمایش شمارش پلاکت و لام خونی: کاهش تعداد و اندازه بزرگ پلاکتها (در ۶۸% موارد).
- تستهای عملکرد پلاکت: مانند تجمع پلاکتی با ریستوستین (RIPA) که در BSS کاهش مییابد.
- سیتومتری: اندازهگیری سطح GPIb-IX-V؛ کاهش آن نشانه قطعی است.
- توالییابی ژنتیکی: شناسایی جهشها.
روشهای نوین مانند NGS (Next-Generation Sequencing) در کودکان، BSS را از ITP (پورپورای ترومبوسیتوپنیک ایدیوپاتیک) تمایز میدهد. در ایران، غربالگری پیش از بارداری توصیه میشود.
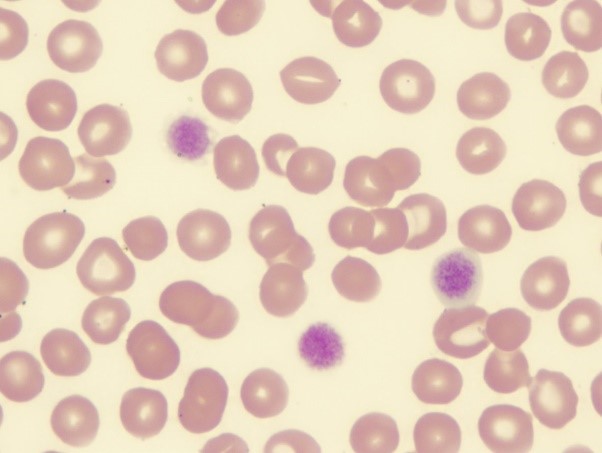
درمان و مدیریت
درمان BSS عمدتاً حمایتی است و بر کنترل خونریزی تمرکز دارد:
- انتقال پلاکت: برای خونریزیهای شدید یا پیش از جراحی؛ مؤثرترین روش.
- داروها: اسید ترانکسامیک (ضدفیبرینولیتیک)، DDAVP برای موارد خفیف و مکملهای آهن برای کمخونی.
- درمان هورمونی: قرصهای ضدبارداری برای منوراژی.
- اجتناب از داروها: آسپیرین و NSAIDها ممنوع هستند.
در موارد اکتسابی، ریتوکسیماب یا درمان عفونت زمینهای مفید است. برای مدیریت perioperative، مانند هیسترکتومی، نظارت دقیق و انتقال پلاکت پیشگیرانه ضروری است.
پیشرفتهای درمانی
ژندرمانی با وکتورهای لنتیویروس، GPIX را در مدلهای سلولی و حیوانی بازسازی کرده و فنوتیپ BSS را اصلاح میکند، اما هنوز در مرحله پیشبالینی است. در هموفیلی و اختلالات مشابه، ژندرمانی پیشرفت کرده که نویدبخش برای BSS است. در ایران، انتقال سلولهای بنیادی خونساز (HSCT) برای موارد شدید پیشنهاد شده، اما دسترسی محدود است.
عوارض و مدیریت در شرایط خاص
عوارض اصلی شامل کمخونی مزمن، خونریزی مغزی نادر و مشکلات بارداری است. در بارداری، خطر خونریزی پس از زایمان افزایش مییابد و نیاز به نظارت هماتولوژیک دارد. پیشگیری با مشاوره ژنتیکی و اجتناب از ضربههای شدید امکانپذیر است. در کودکان، تشخیص زودهنگام از تشخیص اشتباه به عنوان ITP جلوگیری میکند.
نتیجهگیری
سندرم برنارد-سولیر یک چالش ژنتیکی نادر اما قابل مدیریت است که با تشخیص زودهنگام و درمان حمایتی، کیفیت زندگی را بهبود میبخشد. پیشرفتهای مولکولی و ژندرمانی آیندهای روشن ترسیم میکنند. با آگاهی، میتوان از عوارض پیشگیری کرد.
Reference:
https://jmrh.mums.ac.ir/article_23779_041713fafec0d96e9c7944309aa9961a.pdf
https://www.researchgate.net/publication/47642357_A_Study_of_Bernard-Soulier_Syndrome_in_Tehran_Iran
https://jmrh.mums.ac.ir/article_23779.html
https://www.sciencedirect.com/science/article/abs/pii/S0006497124082703
https://pmc.ncbi.nlm.nih.gov/articles/PMC11758589/
https://rarediseases.org/rare-diseases/bernard-soulier-syndrome/
https://pubmed.ncbi.nlm.nih.gov/21039013/
https://www.sciencedirect.com/science/article/pii/S2475037925001979
https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0044-1789184
https://sid.ir/paper/434759/fa
https://civilica.com/doc/2000418/
https://www.apollohospitals.com/fa/diseases-and-conditions/bernard-soulier-syndrome
https://www.beytoote.com/health/malady-remedy/bernard-solier03-syndrome.html
https://arminlab.com/bernard-soulier-syndrome/
https://mendel-lab.com/services/item/627039