
مقدمه
رتینوبلاستوما (Retinoblastoma) شایعترین تومور بدخیم داخل چشمی در کودکان است که از سلولهای نابالغ شبکیه (رتینا) منشأ میگیرد. این بیماری نادر است و معمولاً در کودکان زیر ۵ سال بروز میکند، اما میتواند در سنین بالاتر نیز مشاهده شود. بر اساس آخرین بررسیهای ما تا شهریور سال 1404، پیشرفتهای قابل توجهی در درک ژنتیکی، تشخیص زودرس و درمان این بیماری صورت گرفته است که منجر به نرخ بقای بالا (تا ۹۵% در مراکز پیشرفته) شده است. با این حال، در کشورهای در حال توسعه مانند کشور ما ایران، تأخیر در تشخیص و محدودیت دسترسی به مراقبتهای تخصصی همچنان چالشهای عمدهای هستند. در این مقاله بر اساس منابع معتبر، تمرکز بر جنبههای علائم، شیوع، پاتوفیزیولوژی، اساس ژنتیکی، مشاوره ژنتیک، تشخیص، پروگنوز و درمان خواهد بود.
علائم
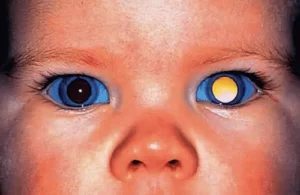
علائم رتینوبلاستوما اغلب در مراحل اولیه بدون علامت هستند، اما با پیشرفت تومور ظاهر میشوند. شایعترین علامت لوکوکوریا یا سفید شدن مردمک چشم است که در حدود ۶۰% موارد مشاهده میشود، به ویژه زمانی که نور (مانند فلاش دوربین) به چشم تابیده میشود. دیگر علائم عبارتند از:
- استرابیسموس (انحراف چشم) یا چپچشمی (در ۲۰-۳۰% موارد).
- کاهش بینایی، که ممکن است با افتادن مکرر کودک یا نزدیک کردن اشیاء به صورت همراه باشد.
- التهاب یا قرمزی چشم، درد، گلوکوم (افزایش فشار داخل چشم) در مراحل پیشرفته.
- در موارد پیشرفته، پروپتوز (جلوآمدگی چشم)، کاهش وزن یا علائم متاستاتیک مانند درد استخوان.
در مطالعات اخیر، تأکید شده که والدین اغلب لوکوکوریا را در عکسهای فلاشدار متوجه میشوند. در ایران، بر اساس مطالعهای در سال ۲۰۰۹ (Naseripour et al.)، تأخیر تشخیص (میانگین ۶-۸ ماه) منجر به علائم پیشرفتهتر میشود. علاوه بر این، علائم کمتر شایع شامل هیتروپی (انحراف عمودی چشم)، ناهماهنگی مردمکها و در موارد نادر، علائم سیستمیک مانند تب یا کمخونی هستند. بررسیهای ۲۰۲۵ نشان میدهد که در کودکان بالای ۵ سال، علائم ممکن است با تومورهای ثانویه مانند پینئوبلاستوما همراه باشد، که نیاز به غربالگری از طریق تصویر برداری عصبی دارد.
جدول علائم شایع رتینوبلاستوما
| علائم | شیوع تقریبی به درصد | توضیحات |
| لوکوکوریا | ۵۰-۶۰ | سفید شدن مردمک، اغلب در عکسها مشاهده میشود. |
| استرابیسموس | ۲۰-۳۰ | انحراف چشم، ممکن است اولین علامت باشد. |
| کاهش بینایی | ۱۰-۲۰ | در کودکان بزرگتر، با مشکلات رفتاری همراه است. |
| قرمزی و التهاب چشم | ۵-۱۰ | نشاندهنده گلوکوم یا نفوذ تومور. |
| پروپتوز | <۵ | در مراحل پیشرفته، با خطر متاستاز. |
| علائم متاستاتیک | <۵ | درد استخوان، کاهش وزن؛ بیشتر در کشورهای کمدرآمد. |
شیوع
شیوع جهانی رتینوبلاستوما ۱ در ۱۶۰۰۰-۱۸۰۰۰ تولد زنده است، با حدود ۸۰۰۰ مورد جدید سالانه. در آسیا، به دلیل عوامل جمعیتی نرخ بالاتر است (تا ۱ در ۱۵۰۰۰). در ایران، بر اساس مطالعات ۲۰۰۱-۲۰۰۷، شیوع ۱.۳۹ مورد جدید در سال به ازای هر میلیون جمعیت است، با نرخ بالاتر موارد دوطرفه (۲۸.۲%). مطالعه GBD نشاندهنده کاهش مرگومیر جهانی اما افزایش موارد در کشورهای با درآمد متوسط (مانند ایران) است. در ۲۰۲۱، شیوع جهانی ۶۲۷۵ مورد تخمین زده شده است. در ایران، نرخ بالاتر ابتلا به این سرطان در جنوب غربی کشور گزارش شده است.
پاتوفیزیولوژی
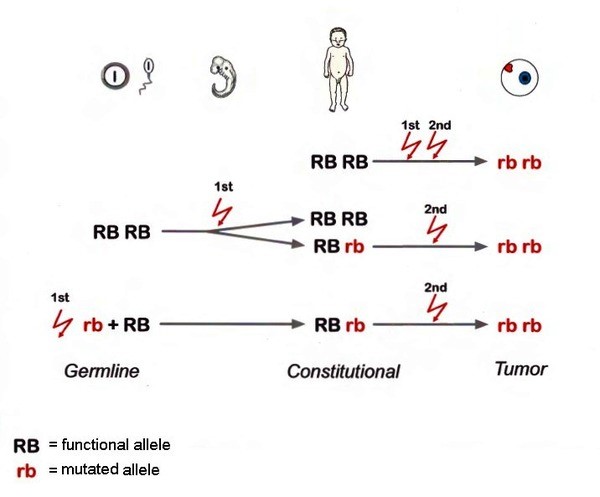
رتینوبلاستوما از جهش در سلولهای پیشساز مخروطی شبکیه ناشی میشود. پاتوفیزیولوژی اصلی این بیماری بر اساس مدل “دو ضربه” (Two-hit hypothesis) نادسون است: در این مدل، غیرفعال شدن هر دو آلل ژن RB1 منجر به از دست رفتن کنترل چرخه سلولی و تکثیر کنترلنشده سلولها میشود. تومور میتواند یکی از 3 حالت زیر باشد:
- اندوفیتیک (رشد به سمت ویتره، با دانهریزی توموری).
- اگزوفیتیک (رشد به سمت فضای زیرشبکیهای).
- یا ترکیبی.
در مراحل پیشرفته، تومور میتواند به عصب بینایی، کوروئید یا خارج چشم نفوذ کند و متاستاز به مغز، استخوان یا غدد لنفاوی بدهد. هیستولوژی تومور شامل سلولهای بازوفیلیک با هستههای هیپرکروماتیک و سیتوپلاسم کم است و ساختارهایی مانند روزتها (Flexner-Wintersteiner یا Homer Wright) نشاندهنده تمایز هستند. عوامل محیطی مانند عفونت HPV در برخی مطالعات پیشنهاد شده، اما نقش اصلی، مربوط به ژنتیک است. پاتوژنزیس همچنین شامل اختلال در مسیر p53 و افزایش بیان ژن MYCN در موارد غیرجهشی RB1 است. مطالعات اخیر نشاندهنده نقش هتروژنی توموری (tumor heterogeneity) در مقاومت به درمان است، جایی که زیرجمعیتهای سلولی با جهشهای اضافی مانند BCOR یا CREBBP باعث پیشرفت بیماری میشوند. علاوه بر این، پاتوفیزیولوژی شامل اختلالات اپیژنتیکی مانند متیلاسیون پروموتر ژن RB1 است که در ۱۰-۱۵% موارد غیرجهشی دیده میشود.
جدول انواع رشد توموری در رتینوبلاستوما
| نوع رشد | ویژگیها | ریسکها |
| اندوفیتیک | رشد به ویتره، دانهریزی | خطر پخش به ویتره و متاستاز CNS |
| اگزوفیتیک | رشد زیرشبکیهای، جدایی شبکیه | خطر نفوذ به کوروئید و عصب بینایی |
| ترکیبی | هر دو ویژگی | پروگنوز ضعیفتر، نیاز به درمان چندمدالیتی |
| دیفوز | پخش یکنواخت، نادر | تشخیص سختتر، مقاومت به درمان |
اساس ژنتیکی
رتینوبلاستوما عمدتاً به دلیل جهش در ژن RB1 (واقع در کروموزوم 13q14) ایجاد میشود که یک ژن سرکوبگر تومور است. انواع ژنتیکی این سرطان عبارتند از:
- ارثی (۴۰-۴۵%): جهش ژرمینال در یک آلل، که اغلب دوطرفه یا چندکانونی است. نرخ نفوذ ۹۰% است و خطر تومورهای ثانویه (مانند سارکوم) را افزایش میدهد.
- غیرارثی (۵۵-۶۰%): جهش سوماتیک در هر دو آلل، معمولاً یکطرفه.
در ۱۰% موارد، موزاییسم ژنتیکی وجود دارد که مشاوره ژنتیکی را پیچیده میکند. مطالعات اخیر نشاندهنده نقش تقویت ژن MYCN است. این نوع جهش در ۱-۳% مواردی که بدون جهش در ژن RB1 هستند، رخ می دهد. در ایران، شیوع جهشهای خاص ژن RB1 مشابه جهانی است، اما مطالعات محدود نشاندهنده نرخ بالاتر موارد ارثی در خانوادههای با سابقه است. تحقیقات در ایران و کنیا بر کشف جهشهای نوین RB1 با استفاده از Sanger sequencing و NGS تمرکز دارد و جهشهایی مانند pR552* را شناسایی کرده که ممکن است کسب عملکرد ایجاد کند. همچنین، نقش ژنهای دیگر مانند BCOR و CREBBP در هتروژنی توموری برجسته شده است.
جدول جهشهای ژنتیکی شایع در رتینوبلاستوما
| نوع جهش | شیوع به درصد | توضیحات |
| جهش نقطهای | ۴۰-۵۰ | اغلب بی معنی یا تغییر قالب، منجر به پروتئین ناقص. |
| حذف بزرگ | ۲۰-۳۰ | حذف بخشی از کروموزوم 13q، مرتبط با موارد ارثی. |
| تقویت ژن MYCN | ۱-۳ | در افراد بدون جهش در ژن RB1، با پروگنوز متفاوت. |
| موزاییسم | ۱۰ | جهش در بخشی از سلولها، تشخیص سختتر. |
| اپیژنتیک / متیلاسیون | ۱۰-۱۵ | افزایش متیلاسیون پروموتر ژن RB1، بدون تغییر در توالی. |
مشاوره ژنتیک
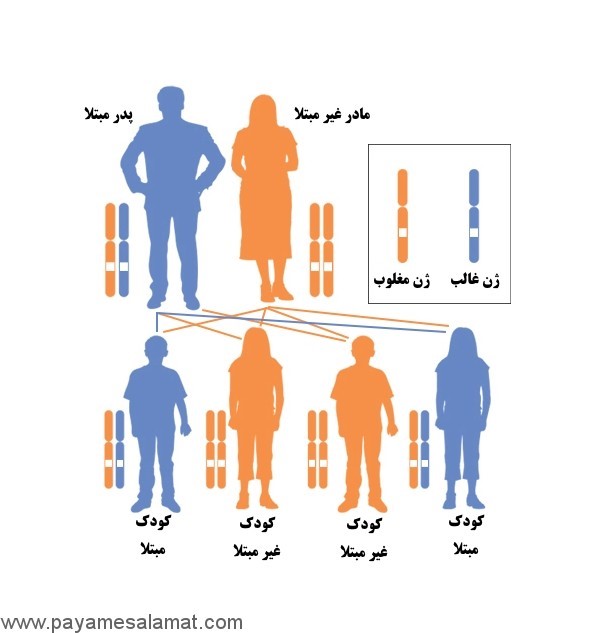
مشاوره ژنتیکی برای خانوادههای مبتلا ضروری است و بر اساس دستورالعملهای NCCN و ICMR شامل موارد زیر میشود:
- ارزیابی خطر ارثی: آزمایش ژنتیکی RB1 برای والدین و خواهر و برادر.
- غربالگری: معاینه چشمی منظم (هر ۳-۴ ماه تا ۵ سالگی) برای کودکان در معرض خطر.
- بحث در مورد خطر تومورهای ثانویه و باروری (مانند ذخیره تخمدان قبل از درمان).
در موارد ارثی، نرخ خطر برای فرزندان ۵۰% است. در ایران، بر اساس دستورالعملهای وزارت بهداشت، مشاوره ژنتیکی در مراکز تخصصی مانند بیمارستان فارابی یا انستیتو کانسر انجام میشود، اما دسترسی محدود است. پیشرفتهای جدید شامل آزمایشهای پیش از تولد و بیوپسی مایع (Liquid Biopsy) برای تشخیص زودرس است. مطالعات ایرانی نشاندهنده اهمیت غربالگری ژنتیکی در ۱۰۶ بیمار، با شناسایی جهش در ۷۳ مورد دو طرفه است. همچنین، دستورالعملهای NCCN ۲۰۲۵ بر غربالگری طولانیمدت برای بازماندگان ارثی تأکید دارد. مشاوره باید شامل بحث در مورد گزینههای باروری مانند PGD باشد.
روشهای تشخیص آزمایشگاهی و ژنتیک
تشخیص اولیه بر اساس معاینه چشمی (با بیهوشی) و تصویربرداری است:
- آزمایشگاهی: بیوپسی معمولاً اجتناب میشود به دلیل خطر پخش تومور، اما در موارد مشکوک، آسپیراسیون ویتره یا مایع قدامی انجام میشود.
- تصویربرداری: اولتراسوند، OCT، MRI برای ارزیابی گسترش (به عصب بینایی یا خارج چشم).
- ژنتیک: آزمایش مولکولی RB1 (Sequencing, MLPA) برای جهشها. در ۹۵% موارد ارثی، جهش شناسایی میشود.
دستورالعملهای NCCN (۲۰۲۵) تأکید بر طبقهبندیICRB (International Classification of Retinoblastoma) برای staging دارند: گروه A (کوچک) تا E (پیشرفته). در ایران، تشخیص اغلب با تأخیر صورت میگیرد و نرخ گسترش خارج چشمی بالاتر است (تا ۵۰% در مطالعات محلی). پیشرفتهای اخیر شامل استفاده از NGS برای تشخیص جهشهای نوین و بیوپسی مایع برای شناسایی ctDNA (circulating tumor DNA) در خون یا مایع زجاجیه است. همچنین، آزمایشهای جدید مانند شناسایی parent-of-origin mutation برای ارزیابی ریسک بالاتر جهش پدری است.
جدول طبقهبندی ICRB برای رتینوبلاستوما
| گروه | پروگنوز | ویژگیها |
| A | >۹۰ | تومور کوچک (<۳ mm)، دور از فووهآ و عصب |
| B | ۸۰-۹۰ | تومور بزرگتر، بدون دانهریزی |
| C | ۷۰-۸۰ | دانهریزی موضعی |
| D | ۴۰-۶۰ | دانهریزی گسترده یا جدایی شبکیه |
| E | <۲۰ | تومور پیشرفته، گلوکوم یا خونریزی |
تعیین پروگنوز
پروگنوز بر اساس مرحله، گسترش و وضعیت ژنتیکی تعیین میشود:
- نرخ بقا: ۹۵% در کشورهای پیشرفته اگر تومور محدود به چشم باشد؛ کاهش به ۶۰% با گسترش عصب بینایی.
- عوامل بد پروگنوز: گسترش خارج چشمی، متاستاز، تأخیر تشخیص، گروه D/E در ICRB.
- حفظ چشم: تا ۹۰% در گروههای اولیه با درمان موضعی.
در مطالعات جهانی، نرخ بقای ۵ ساله در کشورهای کمدرآمد ۵۰% است. در ایران، نرخ بقا حدود ۷۰-۸۰% است، اما حفظ چشم پایینتر (۴۰-۵۰%) به دلیل تشخیص دیررس. عوامل جدید پروگنوز شامل بیان ژنهایی مانند MYCN و هتروژنی توموری است، که با بیومارکرها ارزیابی میشود. همچنین، در کودکان بالای ۵ سال، پروگنوز ضعیفتر به دلیل تشخیص دیرتر.
جدول عوامل پروگنوز در رتینوبلاستوما
| عامل | تأثیر بر پروگنوز | توضیحات |
| مرحله ICRB | گروه A-B: خوب؛ گروه D-E: ضعیف | بر اساس اندازه و گسترش. |
| گسترش خارج چشمی | کاهش بقا به ۵۰% | متاستاز به CNS یا استخوان. |
| وضعیت ژنتیکی | ارثی: خطر ثانویه بالا | نرخ بقا کلی خوب، اما نظارت طولانی. |
| تأخیر تشخیص | کاهش حفظ چشم به ۴۰% | شایع در کشورهای در حال توسعه. |
| درمان اولیه | IAC: حفظ چشم ۸۰% | در مقایسه با انوکلیاسیون. |
درمان
درمان چندمدالیتی و بر اساس مرحله بیماری صورت می گیرد، با هدف حفظ چشم و بینایی:
- شیمیدرمانی: داخل وریدی (IVC) برای موارد سیستمیک (Vincristine, Etoposide, Carboplatin)؛ داخل شریانی (IAC) برای موارد یکطرفه پیشرفته (Melphalan, Topotecan) .
- درمان موضعی: کرایوتراپی، لیزر (TTT)، داخل ویترهای (IvitC با Melphalan) برای دانهریزی.
- پرتودرمانی: پلاک (Brachytherapy) یا EBRT برای موارد مقاوم.
- جراحی: انوکلیاسیون (Enucleation) برای گروه E یا موارد پیشرفته.
دستورالعملهای NCCN (۲۰۲۵)، IAC را به عنوان خط اول برای گروه C-D توصیه میکنند، با نرخ حفظ چشم ۸۰%. در ایران. IVC و انوکلیاسیون شایعتر هستند، اما IAC در مراکز مانند تهران در حال گسترش است. پیشرفتهای اخیر شامل نانودراگها و ایمونوتراپی است. همچنین، chemoplaque (پلاک شیمیدرمانی) برای موارد vitreous seeding، با نرخ موفقیت ۵۹% در حفظ چشم گزارش شده است. درمانهای نوین مانند ویروس های انکولیتیک و ژن درمانی در مراحل آزمایشی هستند.
جدول درمان بر اساس گروه ICRB
| ICRB | درمان اصلی | نرخ حفظ چشم به درصد | توضیحات |
| A | لیزر یا کرایوتراپی | >۹۵ | درمان موضعی کافی. |
| B | IAC یا IVC + لیزر | ۸۵-۹۵ | شیمیدرمانی موضعی اولویت. |
| C | IAC + IvitC | ۷۰-۸۵ | برای دانهریزی موضعی. |
| D | IAC + IVC، ممکن EBRT | ۵۰-۷۰ | خطر انوکلیاسیون بالا. |
| E | انوکلیاسیون + شیمیدرمانی سیستمیک | <۱۰ | تمرکز بر بقا. |
نتیجهگیری
رتینوبلاستوما با تشخیص زودرس و درمان پیشرفته قابل درمان است، اما به نظر می رسد برنامههای غربالگری ملی این بیماری در ایران ضروری است. با پیشرفتهای اخیر، مانند ادغام هوش مصنوعی در تصویربرداری و درمانهای شخصیسازیشده، امید به بهبود بیشتر پروگنوز وجود دارد.
Reference:
https://www.ncbi.nlm.nih.gov/books/NBK659138/
https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1450
https://www.cancer.org/cancer/retinoblastoma.html
https://pubmed.ncbi.nlm.nih.gov/38765425/
https://pubmed.ncbi.nlm.nih.gov/38805607/
https://pubmed.ncbi.nlm.nih.gov/37540503/
https://pubmed.ncbi.nlm.nih.gov/37337407/
https://pubmed.ncbi.nlm.nih.gov/36395983/
https://pubmed.ncbi.nlm.nih.gov/38077901/
https://pubmed.ncbi.nlm.nih.gov/34972201/
https://pubmed.ncbi.nlm.nih.gov/35642324/
https://pubmed.ncbi.nlm.nih.gov/36120973/
https://pubmed.ncbi.nlm.nih.gov/39096073/
https://pubmed.ncbi.nlm.nih.gov/39185198/
https://pubmed.ncbi.nlm.nih.gov/38819901/
https://pubmed.ncbi.nlm.nih.gov/37550664/
https://pubmed.ncbi.nlm.nih.gov/38402833/
https://pubmed.ncbi.nlm.nih.gov/39242324/
https://pubmed.ncbi.nlm.nih.gov/38765425/
https://pubmed.ncbi.nlm.nih.gov/38787654/